
Abstract
Background
Drug resistance is one of the main reasons of anti-malarial treatment failures and impedes malaria containment strategies. As single nucleotide polymorphisms (SNPs) have been found to correlate with anti-malarial drug resistance, the surveillance strategy includes continuous monitoring of known molecular markers and detection of new mutation patterns. With the introduction of artemisinin-based combination therapy, selection of specific patterns has been observed worldwide.
Methods
From March to June 2013, whole blood was collected on filter paper from microscopically malaria positive patients in Jimma zone (District), southwestern Ethiopia. Plasmodium falciparum, Plasmodium vivax and mixed infections were included. SNPs were investigated by conventional or real-time PCR, restriction fragment length pattern analysis or sequencing. Results were compared to molecular patterns from Ethiopian isolates in 2004, 2006 and 2008/9.
Results
Plasmodium falciparum, P. vivax, and mixed infections were molecularly confirmed in 177, 80, and 14 samples, respectively. In P. falciparum, mutations in the pfcrt, pfmdr 1and pfATP 6 (SERCA) gene were investigated. Whereas the mutation in the pfcrt gene at codon 76 K was still found in 95.6 % of all samples, the pfmdr 1 86 T mutation fell to 1.2 % (2/163) in 2013 compared to 9 % in 2008/9 and 86 % in 2006 (P <0.001). The pfmdr 1 184 F mutation dominated with 100.0 % (172/172) in 2013. Sequencing of the recently reported PF3D7_1343700 kelch propeller domain showed no mutation at codon 476. First sequencing data of the pvmdr 1 gene from Jimma region revealed a prevalence of the mutations 976 F and 1076 L in 72.7 % (16/23) and 100.0 % (19/19) of the isolates, respectively.
Conclusion
Since the introduction of artemether-lumefantrine (AL) in Jimma, Ethiopia, in 2006, the prevalence of certain SNPs associated with AL use has increased. Markers for chloroquine resistance in P. vivax were highly frequent. Continuous molecular and clinical surveillance are of paramount importance.
Similar content being viewed by others
Background
The rise of drug-resistant parasites threatens to hamper malaria containment strategies. After the introduction of artemisinin-based combination therapy (ACT) in Jimma zone in 2006 [1], a certain shift in the distribution of wild types and mutations of defined single nucleotide polymorphisms (SNPs), associated with anti-malarial drug resistance, was expected.
The Plasmodium falciparum chloroquine resistance transporter (pfcrt) gene on chromosome 7 encodes a transmembrane protein to be found in the digestive vacuole of the parasite and has primarily been associated with resistances to chloroquine (CQ). However, it also seems to influence artemisinin, quinine and amodiaquine susceptibility [2–4]. The P. falciparum multi-drug resistance protein 1 (pfmdr 1) gene, located on chromosome 5, encodes a P-glycoprotein homologue 1 [5, 6]. Point mutations in codon 86 and 184 seem to be involved in artemether-lumefantrine (AL) resistance in some [7–10] but not all studies [11]. Concerning CQ, the PfMDR 1 protein is believed to add a modulatory effect to resistance mechanisms without conferring actual resistance [12]. An increased copy number has been linked to affect mainly mefloquine, but also artesunate, dihydro-artemisinin, halofantrine, quinine, and lumefantrine susceptibility [13–15]. Increased copy numbers of this gene however, do appear to play a minor role on the African continent [8, 9, 16].
The P. falciparum SERCA-type ATPase 6, encoded by the pfATP 6 gene, was suggested to be the cytosolic target structure of artemisinins [17–19]. A variety of polymorphisms located in the pfATP 6 gene have been reported [20–23]. However, their actual importance concerning artimisinin resistance remains unclear. Recently, mutations in the K13-propeller gene encoding the PF_1343700 kelch propeller domain have been proposed to be determinants for artemisinin resistance [24]. Yet its actual function within the parasite organism is unknown and can only be assumed, as homologous proteins in other organism can be found participating in a wide variety of pathways [24–26].
The P. vivax multi-drug resistance protein1, encoded by the pvmdr 1 gene, is the pfmdr 1 orthologue in P. vivax and is presumed to be connected to changes of CQ, amodiaquine and sulfadoxine-pyrimethamine treatment response. Specifically mutations in codon 976 and 1076 seem to be responsible for the aforementioned associations [27–29].
In this present study, the prevalence of these molecular markers in southwestern Ethiopia collected in 2013 was compared to earlier published and partially unpublished data from 2004, 2006 and 2009 from this region [30–32].
Methods
Study site and patient material
Sample collection took place from March until June 2013 in five different health centres in Goma province around Jimma town, approximately 355 km southwest of the Ethiopian capital Addis Ababa. Malaria transmission is seasonal with minor transmission from April to June and major transmission from September to December. The study was implemented in the routine diagnostic settings. When malaria was clinically suspected, microscopic investigations of blood slides were performed. Slides were stained with Giemsa and independently read by two experienced microscopists. Only microscopically malaria-positive patients were considered for inclusion in the study. Plasmodium falciparum and P. vivax single infections as well as mixed infections were included in the study. Pregnant women and children under the age of one year were excluded. Ethical clearance was granted by the Ethical Committee of the Universitiy of Jimma, Ethiopia, as well as of the University of Munich, Germany. A signed and dated informed consent form was obtained from each patient or parent/guardian as applicable. Blood samples for molecular investigations were derived from fingerprick samples applied on filter paper (Standard-Whatman Cellulose Chromatography paper 3MM, GE Healthcare, Fairfield, CT, USA) after recruitment. Filter papers were air-dried, individually packed in sterile DNA free zipper bags and stored in Jimma at ambient temperature. After arrival in Munich, samples were stored frozen at −20 °C.
DNA extraction and amplification
DNA was extracted from filter paper using the Chelex method as described earlier [31, 33]. For parasite specification, a nested conventional polymerase chain reaction (PCR) was used [34]. In order to survey the pfcrt 76 K mutation, a nested conventional PCR followed by enzyme digestion with ApoI was performed as described previously [2, 35]. The specific restriction fragment length pattern (RFLP) analysis enables to discriminate between mutation and wild type. The validity of the assay was confirmed by sequencing 10 % of the samples and comparing the obtained results with the RFLP result.
To analyse pfmdr 1 mutations at codons 86 and 184, the real-time PCR described by Purfield et al. was established [36]. Again, the validity of the method was confirmed by sequencing of 10 % of the samples. Pfmdr 1 copy numbers were investigated as previously described in more detail [14]. The method was thereby adapted for a Biorad real-time PCR cycler.
Gel electrophoresis was performed with DNA GelRed™ stained 1.25-2 % agarose gels. For RFLP discrimination, high-resolution agarose (Roth®) was used.
Sequencing was performed using Big Dye Terminator 3.1 and DyeEx® 2.0 Spin Kit as described earlier [31]. Amplified fragments from pfATP 6, pvmdr 1 and the newly described K13-propeller gene were sequenced using the same primer sequences as used for PCR amplification. For all primer sequences refer to Additional file 1. Genomic sequences were analysed by the sequence alignment editors Bioedit and DNASIS MAX (version 3.0). Reference sequences were obtained from the National Center for Biotechnology Information (NCBI). Sequence similarity was investigated via Basic Local Alignment Search Tool (BLAST) [37]. (Reference sequences: pfmdr 1 3D7 AL844501.1, pfcrt XM_001348968.1, pfATP 6 AB121059.1, K13 propeller XM_001350122.1, pvmdr 1 EU333975.1,).
Results
Sample collection and species differentiation
Altogether 338 finger prick samples of microscopically positive malaria patients were collected. Seventeen (5.0 %) samples originated from Choche Health Centre, 24 (7.1 %) from Limu Shay Health Centre, 154 (45.6 %) from Didisa Health Centre, three (0.9 %) from Jimma Health Centre, and 140 (41.4 %) from Shanan Gibe Hospital. Plasmodium falciparum and P. vivax could be detected by microscopy in 197 (61.2 %) and 98 (30.4 %) samples, respectively. A total of 27 (8.4 %) samples were classified as mixed infections with both species. For 16 samples, microscopic data were missing.
Molecular species differentiation by nested PCR confirmed 177 samples of P. falciparum, 80 samples of P. vivax and 14 mixed samples. A 67 of 338 (19.8 %) samples remained molecularly malaria-negative. Repetition and increase in DNA template resulted in the same result. Prevalence of P. falciparum and P. vivax mono-infections in these 271 positive samples were 65.3 % and 29.5 %, respectively.
Molecular patterns in Plasmodium falciparum
In P. falciparum the following codons were assessed, mixed infections were included in the analysis: pfcrt 76, pfmdr1 86 and 184, pfATP 6 codons 263–431 and the recently published K13-propeller domain with codon 476. The amplification and enzyme digestion of the pfcrt amplicon was successful for 159 samples. Within these, 152 (95.6 %) samples showed the mutation at codon 76 T, whereas only seven showed wild type (76 K) characteristics (Table 1). No mixed infections (wild type and mutation in one sample) could be detected. Sequencing 10 % of the samples revealed that both, the CVIET and CVMNT haplotype are present in 50 % of cases in codons 72–76. Interestingly, in 2009 the CVIET haplotype could be found in all cases.
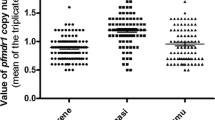
Concerning the pfmdr 1 gene, only two samples were identified as pfmdr 1 86Y mutants. The prevalence of the 86 N dominated with 98.8 % (n = 161). The pfmdr 1 Y184F haplotype was found in all 171 amplified samples (Table 1). No mixed infections were observed. Pfmdr copy numbers were only investigated in 2009, all samples showed only one copy of the gene.
Sequencing of the pfATP 6 gene was performed from 50 randomly chosen samples, the K13- propeller region was sequenced from 25 randomly selected samples. The pfATP 6 431 K mutation showed a prevalence of 18.8 % (9/48). One sample failed to amplify properly, another sample was not distinguishable by sequencing results (Table 1). Given that the sequences often were too short to allow full judgement, no other mutations could be confidently proven.
As the K13-propeller gene is uncommonly long for the sequencing method used, only sequences from codon 343 to codon 693 could reliably be assessed. No mutations including M476I as described in Ariey et al. [24] were detected. However, one mutation at codon 531 (N531I) was identified that had not been described earlier (Table 1).
There was no significant difference in the distribution of molecular patterns between the different health centres.
Molecular patterns in Plasmodium vivax
Sequencing results of the pvmdr1 gene have not been reported before from this region.
From 24 out of 25 randomly chosen samples, a partial pvmdr1 sequence could be successfully amplified and sequenced. The homology with the reference strain (EU333975.1) was 100 % for the wild type. The sequence proved to be highly conserved on a global level. There was no difference in similarity between regionally closer isolates compared to isolates from other continents apart from the formerly described mutations at codon 976 and 1076. Prevalence of the Y976F mutation was 72.7 %, all readable samples showed the F1076L mutation. Two and five samples were indistinguishable for codon 976 and 1076, respectively. No other mutations were found in these sequences spanning from codon 938 to 1086. There was no significant difference in the distribution of molecular patterns between the different health centres. Two exemplary sequences are published at GenBank [38].
Mutations over time
Mutations in the following P. falciparum genes were investigated over time: pfcrt, pfmdr, and pfATP 6 (Fig. 1). The sample size was 98 and 251 in 2006 and 2009, respectively. Mutation analysis of the pfcrt and pfmdr 1 gene was performed for all samples. Confirmative sequencing was conducted for 10 % of these samples in 2009. On average, 15 % and 10 % of all samples were successfully sequenced at the pfATP 6 gene in 2006 and 2009, respectively.

Prevalence of investigated mutations over time. Alterations of molecular markers with regard to the introduction of ACT in Jimma zone in 2006. No significant change in the pfcrt gene sequence was observed. The SERCA 431 K mutation dropped from 58.3 % in 2006 to 18.8 % in 2013 but the sample size was very low. Changes in the pfmdr 1 gene were remarkable. In 2006 the mutation at codon 86Y was dominant with 80.6 %. Mixed infections were taken together with wild type samples. The wild type alone was only detectable in 14.3 %. In 2009 and 2013, only 9.2 % and 1.2 % showed the mutation 86Y, respectively. The prevalence of pfmdr 1 84 F was high in 2009 and remained dominant. Data from 2004 are taken from reference 30
There was no significant change in the pfcrt gene observed. Prevalence of the wild type remained negligible (4.4 %). However, changes in the pfmdr 1 gene were remarkable. Until 2006, the mutation at codon 86Y was dominant: 80.6 % (79/98) of the samples showed the mutation, only 14.3 % presented the wild type, the rest were mixed clone infections presenting both haplotypes [31]. In 2009, the distribution had changed dramatically. Only 9.2 % showed the mutation 86Y and 228 of the 251 samples (90.8 %) presented the wild type 86 N [32]. In 2013, the prevalence of 86Y dropped to as low as 1.2 %. Pfmdr 1 184 F was first investigated in 2009, the prevalence was 98 %, increasing to 100 % in 2013. The pfATP 6 431 K mutation dropped from 58.3 % (7/12) in 2006 to 18.8 % (9/48) in 2013 but the sample size was very low. Other mutations described in this short sequence were not detectable.
Discussion
In this study, molecular resistance patterns in Ethiopian P. falciparum and P. vivax isolates were investigated. Pfcrt 76 T, associated mainly with CQ resistance, could be found almost as frequently as before introduction of ACT. Either continuous CQ use remains common in Jimma region, or the pfcrt wild type provides no fitness advantage under AL treatment. As the wild type 76 K re-emerged under ACT and in absence of CQ in some countries [39], continuous CQ treatment in Jimma region should be at least suspected. CQ is not recommended nor released in the health centres for P. falciparum in Jimma region but available on the free market and recommended for P. vivax treatment.
As shown above, microscopic diagnosis was not always reliable; performance was similar in all health centres. Only in 203 (60.1 %) of all samples, the microscopy result matched the PCR result. A total of 16 samples microscopically diagnosed as vivax malaria could be proven to be P. falciparum infections by PCR. Overall, 19.8 % microscopically positive samples were negative by PCR, 21 were microscopically categorized as vivax malaria and treated accordingly. Therefore, P. falciparum parasites still have opportunities to get in contact with CQ. Also, self-treatment of the rural population in less severe cases seems to be still very common as CQ is cheap and easily available. The prevalence of P. vivax in Jimma region is between 30-70 % [40]. The performance data are consistent with a recent study from Ethiopia assessing malaria diagnostic capacities in Ethiopian health centres [41]. A recent study from Mozambique showed a very similar changing pattern. The pfmdr 1 allele N86 increased from 19.5 % in 2003–2005 to 73.2 % in 2010–2012 after introduction of AL [42]. Interestingly, a recent study from southeast Ethiopia (Omo Nada, Bala Wajo, Arba Minch and Harar) showed different results [43]. The authors declared a consequent absence of CQ use in this area. Prevalence of 76 T was 13.5 % (23/170) in the South and 32 % (8/25) in the East. Overall, the C72S mutation was observed only in 3.6 %.
The authors further stated that the CQ-sensitive CVMNK haplotype was found in 95.9 %, the mutant haplotype SVMNT in 4.1 % and CVIET was absent at codons 72–76. Sequencing of the mutant samples from Jimma showed in 100 % the CVIET haplotype in 2009 and 50 % CVIET / 50 % CVMNT in 2013, in accordance with other studies from Africa [32]. The C72S mutation was absent as well as the sensitive CVMNK haplotype. However, a shift to CVMNT might be a possible interpretation. These regional differences are highly interesting and warrent further investigation.
The Pfmdr 1 wild type 86 N was frequently detected in areas where AL is used. A selection for or re-introduction of the wild type is discussed for artemether as well as lumefantrine and might be the explanation for this drastic change [7–10]. Since the introduction of AL in Jimma zone in 2006, the prevalence of the wild type remarkably increased (P <0.001, data from 2006 compared to 2013 by Wilcoxon rank-sum test; Fig. 1). Comparing the pfmdr1 184 haplotype with data from 2009, the prevalence of the mutation remained stable at above 98 %. These findings support the theory that the pfmdr1 184 F is selected under drug pressure, especially under AL treatment [9]. Unfortunately, no earlier data exists for pfmdr 1 Y184F from this area. A recent study from southeast Ethiopia reported a prevalence of 9 % for N86 [40]. This is similar to the data observed in this study. The prevalence of Y184F was 5 % only compared to 100 % in this study. Different selection mechanisms might be responsible. The diagnostic tool used in this study consisted of a real-time PCR with selective probes, sequencing was only performed in 10 % of the samples but the latter consistently confirmed the results of the PCR.
In 2009, all samples from Jimma showed only one pfmdr copy. In the above-mentioned study from southeast Ethiopia, all samples presented also with only one copy [43]. This is in accordance with other studies from Africa [8, 9, 16].
The prevalence of the E431K mutation in the pfATP 6 gene dropped from 58.3 % in 2006 [31] to 18.8 % in 2013. Recently, E431K together with A623E has been described to reduce artimisinin susceptibility [20]. The A623E mutation was extremely rare with 1/24 (4.2 %) in 2006 and 0/33 (0 %) in 2009. No samples with both mutations could be detected. The mutation was not assessed in 2013 but the decline of the E431K mutation contradicts selection of it under artemisinin pressure.
The new potential candidate referring resistance to artemisinins, the K13 propeller gene, showed no mutations at codon 476. This mutation has been described by Ariey et al. in highly resistant samples from Cambodia [24]. A new mutation at codon N531I was found in only one isolate. Clinical effectiveness of AL was still 95 % in 2009. Clearance rates showed no significant prolongation, no resistance could be suspected then. More recent clinical data from this area were not available [40].
Sequencing of the pvmdr 1 gene revealed a high prevalence of the Y976F and F1076L mutations. This correlates well with the long lasting CQ use in this area. Treatment failures were reported in recent P. vivax studies [43–46]. Comparison of the Ethiopian pvmdr 1 sequences to sequences from Brazil (AY571984.1), Cambodia (JQ925836.1), India (KC818412.1), Korea (GU476519.1, GU244390.1) Madagascar (EU683815.1) and Thailand (KC121338.1) showed highly conserved features without systematic geographical clustering.
Conclusion
The prevalence of SNPs associated with drug resistance has been influenced since the introduction of AL treatment in Ethiopia in 2006 (P <0.001). The wild type of pfmdr codon 86 seems to possess a selection advantage. No significant change was observed for the pfcrt codon 76. First sequencing data of the K13 propeller region revealed no mutations as described in Asia. Mutations associated with CQ resistance in pvmdr 1 were highly prevalent. Continuous molecular and clinical surveillance are of paramount importance.
References
Gürkov R, Eshetu T, Miranda IB, Berens-Riha N, Mamo Y, Girma T, et al. Ototoxicity of artemether/lumefantrine in the treatment of falciparum malaria: a randomized trial. Malar J. 2008;7:179.
Djimdé A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S, Diourté Y, et al. A molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med. 2001;344:257–63.
Schneider AG, Premji Z, Felger I, Smith T, Abdulla S, Beck HP, et al. A point mutation in codon 76 of pfcrt of P. falciparum is positively selected for by Chloroquine treatment in Tanzania. Infect Genet Evol. 2002;1:183–9.
Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298:210–3.
Foote SJ, Thompson JK, Cowman AF, Kemp DJ. Amplification of the multidrug resistance gene in some chloroquine-resistant isolates of P. falciparum. Cell. 1989;57:921–30.
Cowman AF, Karcz S, Galatis D, Culvenor JG. A P-glycoprotein homologue of Plasmodium falciparum is localized on the digestive vacuole. J Cell Biol. 1991;113:1033–42.
Sisowath C, Strömberg J, Mårtensson A, Msellem M, Obondo C, Björkman A, et al. In vivo selection of Plasmodium falciparum pfmdr1 86 N coding alleles by artemether-lumefantrine (Coartem). J Infect Dis. 2005;191:1014–7.
Sisowath C, Ferreira PE, Bustamante LY, Dahlström S, Mårtensson A, Björkman A, et al. The role of pfmdr1 in Plasmodium falciparum tolerance to artemether-lumefantrine in Africa. Trop Med Int Health. 2007;12:736–42.
Dokomajilar C, Nsobya SL, Greenhouse B, Rosenthal PJ, Dorsey G. Selection of Plasmodium falciparum pfmdr1 alleles following therapy with artemether-lumefantrine in an area of Uganda where malaria is highly endemic. Antimicrob Agents Chemother. 2006;50:1893–5.
Happi CT, Gbotosho GO, Folarin OA, Sowunmi A, Hudson T, O’Neil M, et al. Selection of Plasmodium falciparum multidrug resistance gene 1 alleles in asexual stages and gametocytes by artemether-lumefantrine in Nigerian children with uncomplicated falciparum malaria. Antimicrob Agents Chemother. 2009;53:888–95.
Imwong M, Dondorp AM, Nosten F, Yi P, Mungthin M, Hanchana S, et al. Exploring the contribution of candidate genes to artemisinin resistance in Plasmodium falciparum. Antimicrob Agents Chemother. 2010;54:2886–92.
Roepe PD. Molecular and physiologic basis of quinoline drug resistance in Plasmodium falciparum malaria. Future Microbiol. 2009;4:441–55.
Pickard AL, Wongsrichanalai C, Purfield A, Kamwendo D, Emery K, Zalewski C, et al. Resistance to antimalarials in Southeast Asia and genetic polymorphisms in pfmdr1. Antimicrob Agents Chemother. 2003;47:2418–23.
Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. The Lancet. 2004;364:438–47.
Price RN, Uhlemann AC, van Vugt M, Brockman A, Hutagalung R, Nair S, et al. Molecular and pharmacological determinants of the therapeutic response to artemether-lumefantrine in multidrug-resistant Plasmodium falciparum malaria. Clin Infect Dis. 2006;42:1570–7.
Holmgren G, Björkman A, Gil JP. Amodiaquine resistance is not related to rare findings of pfmdr1 gene amplifications in Kenya. Trop Med Int Health. 2006;11:1808–12.
Eckstein-Ludwig U, Webb RJ, Van Goethem ID, East JM, Lee AG, Kimura M, et al. Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003;424:957–61.
Krishna S, Uhlemann AC, Haynes RK. Artemisinins: mechanisms of action and potential for resistance. Drug Resist Updat. 2004;7:233–44.
Krishna S, Woodrow CJ, Staines HM, Haynes RK, Mercereau-Puijalon O. Re-evaluation of how artemisinins work in light of emerging evidence of in vitro resistance. Trends Mol Med. 2006;12:200–5.
Jambou R, Legrand E, Niang M, Khim N, Lim P, Volney B, et al. Resistance of Plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6. Lancet. 2005;366:1960–63.
Menegon M, Sannella AR, Majori G, Severini C. Detection of novel point mutations in the Plasmodium falciparum ATPase6 candidate gene for resistance to artemisinins. Parasitol Int. 2008;57:233–5.
Jambou R, Martinelli A, Pinto J, Gribaldo S, Legrand E, Niang M, et al. Geographic structuring of the Plasmodium falciparum sarco(endo)plasmic reticulum Ca2+ ATPase (PfSERCA) gene diversity. PLoS One. 2010;5:e9424.
Zakeri S, Hemati S, Pirahmadi S, Afsharpad M, Raeisi A, Djadid ND. Molecular assessment of atpase6 mutations associated with artemisinin resistance among unexposed and exposed Plasmodium falciparum clinical isolates to artemisinin-based combination therapy. Malar J. 2012;11:373.
Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–5.
Adams J, Kelso R, Cooley L. The kelch repeat superfamily of proteins: propellers of cell function. Trends Cell Biol. 2000;10:17–24.
Prag S, Adams JC. Molecular phylogeny of the kelch-repeat superfamily reveals an expansion of BTB/kelch proteins in animals. BMC Bioinformatics. 2003;4:42.
Brega S, Meslin B, de Monbrison F, Severini C, Gradoni L, Udomsangpetch R, et al. Identification of the Plasmodium vivax mdr-like gene (pvmdr1) and analysis of single-nucleotide polymorphisms among isolates from different areas of endemicity. J Infect Dis. 2005;191:272–7.
Suwanarusk R, Russell B, Chavchich M, Chalfein F, Kenangalem E, Kosaisavee V, et al. Chloroquine resistant Plasmodium vivax: in vitro characterisation and association with molecular polymorphisms. PLoS One. 2007;2:e1089.
Marfurt J, de Monbrison F, Brega S, Barbollat L, Müller I, Sie A, et al. Molecular markers of in vivo Plasmodium vivax resistance to amodiaquine plus sulfadoxine-pyrimethamine: mutations in pvdhfr and pvmdr1. J Infect Dis. 2008;198:409–17.
Schunk M, Kumma WP, Miranda IB, Osman ME, Roewer S, Alano A, et al. High prevalence of drug-resistance mutations in Plasmodium falciparum and Plasmodium vivax in southern Ethiopia. Malar J. 2006;5:54.
Eshetu T, Berens-Riha N, Fekadu S, Tadesse Z, Gürkov R, Hölscher M, et al. Different mutation patterns of Plasmodium falciparum among patients in Jimma University Hospital, Ethiopia. Malar J. 2010;9:226.
Venkatesan M, Gadalla NB, Stepniewska K, Dahal P, Nsanzabana C, Moriera C, et al. Polymorphisms in Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes: parasite risk factors that affect treatment outcomes for P. falciparum malaria after artemether-lumefantrine and artesunate-amodiaquine. Am J Trop Med Hyg. 2014;91:833–43.
Kain KC, Lanar DE. Determination of genetic variation within Plasmodium falciparum by using enzymatically amplified DNA from filter paper disks impregnated with whole blood. J Clin Microbiol. 1991;29:1171–4.
Snounou G, Viriyakosol S, Jarra W, Thaithong S, Brown KN. Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol Biochem Parasitol. 1993;58:283–92.
Berens N, Schwoebel B, Jordan S, Vanisaveth V, Phetsouvanh R, Christophel EM, et al. Plasmodium falciparum: correlation of in vivo resistance to chloroquine and antifolates with genetic polymorphisms in isolates from the south of Lao PDR. Trop Med Int Health. 2003;8:775–82.
Purfield A, Nelson A, Laoboonchai A, Congpuong K, McDaniel P, Miller RS, et al. A new method for detection of pfmdr1 mutations in Plasmodium falciparum DNA using real-time PCR. Malar J. 2004;3:9.
Basic Local Alignment Search Tool (BLAST), National Center for Biotechnology Information [http://www.ncbi.nlm.nih.gov/Blast.cgi]
GenBank. NIH genetic sequence database. National Center for Biotechnology Information [http://www.ncbi.nlm.nih.gov/genbank]
WHO. Global report on antimalarial drug efficacy and drug resistance: 2000—2010. Geneva, World Health Organization, 2010. [http://whqlibdoc.who.int/publications/2010/9789241500470_eng.pdf]
Eshetu T, Abdo N, Bedru KH, Fekadu S, Wieser A, Pritsch M, et al. Open-label trial with artemether-lumefantrine against uncomplicated Plasmodium falciparum malaria three years after its broad introduction in Jimma Zone, Ethiopia. Malar J. 2012;11:240.
Abreha T, Alemayehu B, Tadesse Y, Gebresillassie S, Tadesse A, Demeke L, et al. Malaria diagnostic capacity in health facilities in Ethiopia. Malar J. 2014;13:292.
Lobo E, de Sousa B, Rosa S, Figueiredo P, Lobo L, Pateira S, et al. Prevalence of pfmdr1 alleles associated with artemether-lumefantrine tolerance/resistance in Maputo before and after the implementation of artemisinin-based combination therapy. Malar J. 2014;13:300.
Mekonnen SK, Aseffa A, Berhe N, Teklehaymanot T, Clouse RM, Gebru T, et al. Return of chloroquine-sensitive Plasmodium falciparum parasites and emergence of chloroquine-resistant Plasmodium vivax in Ethiopia. Malar J. 2014;13:244.
Teka H, Petros B, Yamuah L, Tesfaye G, Elhassan I, Muchohi S, et al. Chloroquine-resistant Plasmodium vivax malaria in Debre Zeit, Ethiopia. Malar J. 2008;7:220.
Ketema T, Bacha K, Birhanu T, Petros B. Chloroquine-resistant Plasmodium vivax malaria in Serbo town, Jimma zone, south-west Ethiopia. Malar J. 2009;8:177.
Yohannes AM, Teklehaimanot A, Bergqvist Y, Ringwald P. Confirmed vivax resistance to chloroquine and effectiveness of artemether-lumefantrine for the treatment of vivax malaria in Ethiopia. Am J Trop Med Hyg. 2011;84:137–40.
Acknowledgements
We thank the health personnel in all Health Care Centers that were involved in the study and the staff of the parasitological Department at Jimma University Hospital for support and assistance and participating patients for their cooperation. Financial support was received from the University of Munich.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AH carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript. NA and AZ participated in the sample collection and coordination in Ethiopia. TE participated in design and coordination of the study. TL participated in design of the study and manuscript preparation. AW participated in sample collection and drafting of the manuscript. MP participated in coordination in Germany and drafting of the manuscript. NBR conceived of and designed the study, supervised the molecular studies and sequence alignment coordination and drafted the manuscript. All authors read and approved the final manuscript.
Additional file
Additional file 1:
Primers used in the study.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Heuchert, A., Abduselam, N., Zeynudin, A. et al. Molecular markers of anti-malarial drug resistance in southwest Ethiopia over time: regional surveillance from 2006 to 2013. Malar J 14, 208 (2015). https://doi.org/10.1186/s12936-015-0723-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-015-0723-2