
Abstract
Platelets, involved in the whole process of tumorigenesis and development, constantly absorb and enrich tumor-specific substances in the circulation during their life span, thus called “Tumor Educated Platelets” (TEPs). The alterations of platelet mRNA profiles have been identified as tumor markers due to the regulatory mechanism of post-transcriptional splicing. Small nuclear RNAs (SnRNAs), the important spliceosome components in platelets, dominate platelet RNA splicing and regulate the splicing intensity of pre-mRNA. Endogenous variation at the snRNA levels leads to widespread differences in alternative splicing, thereby driving the development and progression of neoplastic diseases. This review systematically expounds the bidirectional tumor-platelets interactions, especially the tumor induced alternative splicing in TEP, and further explores whether molecules related to alternative splicing such as snRNAs can serve as novel biomarkers for cancer diagnostics.
Similar content being viewed by others


Introduction
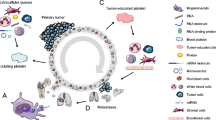
Platelets, the most abundant anucleate cells except red blood cells in the circulation, originate from megakaryocytes in the bone marrow with a short average lifespan of 7 days [1]. Besides its role in hemostasis, platelets also play an important role in tumorigenesis and tumor progression [2]. Platelets stimulate tumor angiogenesis and vascular remodeling, protect CTCs from shear forces and evade immune surveillance, and recruit stromal cells to facilitate the establishment of metastatic niches and promote the metastasis. On the other point of view, tumor can also “educate” platelets. It induces platelet activation, aggregation, and release of platelet-derived substances in circulation, and promote thrombocytosis via influence megakaryopoiesis in bone marrow (Fig. 1). During the bidirectional tumor-platelet interactions, platelets systematically and locally respond to cancer, as well constantly absorb and enrich free proteins, nucleic acids, vesicles and particles [3, 4], leading to the alterations in their RNA and proteomics expression profiles [5, 6], thus termed “tumor educated platelets” (TEPs) [2].

The crosstalk between cancer and platelets. Tumor educates platelets: tumor can induce platelet activation, aggregation, and release of platelet-derived substances in circulation, and promote thrombocytosis via influencing megakaryopoiesis in bone marrow; Platelets support tumor growth and metastasis: platelets stimulate tumor angiogenesis and vascular remodeling, protect CTCs from shear forces and evade immune surveillance, and recruit stromal cells to facilitate the establishment of metastatic niches and promote the metastasis (MKP: megakaryocyte progenitor; MK: megakaryocyte; HSC: Hematopoietic stem cells)
The changes of TEPs profile represent a massive, concentrated biorepository of tumor-derived and bioactive molecules, indicating the potential of TEPs as specific biomarkers for cancer. Due to the short lifespan and the structure of platelet membrane, tumor-specified biosources and biomolecules are enriched in TEPs and protected from circulating RNAase and other enzymes, thus contents in TEP are capable to reflect tumor bioactivity up-to-date, intensive, and dynamically, playing the crucial roles in cancer detection and progression monitoring including colorectal carcinoma (CRC), glioblastoma, non–small cell lung cancer (NSCLC), prostate cancer, and etc. Platelet lacks a nucleus; no genomic DNA is available for transcription of new RNA molecules. Quantification of platelet RNA demonstrates approximately ~ 2.2 fentogram of RNA in one single platelet, but 20–40 times in younger, reticulated platelet [7, 8], indicating a variety of RNA regulatory biological processes, such as RNA splicing.
RNA splicing in TEPs can be induced by external signals (such as platelet surface receptor activation), or in response to signals released by tumor microenvironment, resulting in highly dynamic mRNA repertoires with potential tumor diagnostic applications [9]. Platelets contain many proteins associated with the spliceosome and small nuclear RNAs (snRNAs) to form small nuclear ribonucleoproteins (snRNPs) [10, 11]. SnRNAs including U1, U2, U4, U5, U6 are not merely the basal factors ubiquitously expressed in all cells since they are required for the guidance of pre-mRNA splicing [12], whereas they are extremely variable across a wide range of biological conditions [13]. The endogenous alterations in TEP snRNAs can modulate alternative splicing [14], thereby contributing to the alternation of TEP mRNA profile significantly. Although TEP mRNA has been well-recognized as the promising biomarkers for liquid biopsy in various tumors in recent years [15], it is generally uninformed about the regulation of TEP alternative splicing and its role in cancer diagnostics. This review systematically expounds the bidirectional tumor-platelet interactions, especially the tumor induced alternative splicing in TEP, and further explores whether molecules related to alternative splicing such as snRNAs can serve as novel biomarkers for cancer diagnostics.
The interactions between platelets and tumor
Tumor cells changes platelets
Structure basis of tumor-platelet direct interactions
Direct surface receptor binding and extracellular protein-mediated receptor bridging were the structure basis of tumor-platelet interactions [16,17,18] (Fig. 2). Numerous studies have investigated the targeting direct molecule contacts, including platelet GPIIb-IIIa (also called αIIbβ3 integrin)-plasma fibrinogen or fibronectin - tumor αVβ3 integrin [19,20,21]; platelet GPIbα - tumor Von Willebrand Factor (vWF) [22,23,24]; platelet GPVI - tumor fibrin and/or subendothelial collagen [25, 26]; platelet α6β1 integrin-tumor ADAM9 [27]; platelet acid sphingomyelinase (Asm) – tumor α6β1 integrin [28, 29]; platelet CLEC-2-tumor podoplanin [30,31,32]; and platelet P-selectin-tumor P-selectin ligand [33,34,35]. These platelet receptors and their ligands mediate tumor growth, metastasis and direct tumor-platelet interactions.

Structure basis of tumor-platelet interaction and molecular mechanisms of TCIPA. Direct surface receptor binding and extracellular protein-mediated receptor bridging are the structure basis of tumor-platelet interactions (left); The interactions trigger platelet activation and degranulation, in turn aggregation (TCIPA) dependent on GPIIb-IIIa and fibrin network (right)
Tumor cells induced aggregation
Additionally, tumors can induce platelet aggregation by directly interacting with platelets. Once tumor cells leave the primary tumor site and enter the blood circulation, they directly lead to platelet activation and aggregation, whereby platelets protect tumor cells from immune cell-induced cell death [36], a phenomenon known as “Tumor cell-induced platelet aggregation (TCIPA) [37, 38] " (Fig. 2). In this way, TCIPA can trigger platelets to release a large amount of pro-tumorigenic factors to fuel tumor growth [39]. Current studies have suggested that TCIPA mainly works through the following pathways: (i) tumor cell-platelets interactions result in the formation of small amounts of thrombin, which may trigger platelet activation and aggregation, (ii) fibrinogen binding to integrin αIIbβ3 and fibrin formation can mediate platelet aggregation, and (iii) tumor cells cause some ATP/ADP to be released from dense granules, and the release of ADP stimulates P2Y12 receptors that are necessary for platelet aggregation [37, 40].
Tumor cells promote thrombocytosis
As early as the 19th century, studies first reported the relationship between thrombocytosis and tumors, which was common in tumor patients [41], whereas the interaction of platelets and cancer cells formed a positive feedback cascade to potentiate the effect. It was reported the increased platelet count was associated with poor overall and/or progression-free survival and revealed as predictors of a variety of cancers [42, 43], including lung cancer [44], ovarian cancer [45], gastric cancer [41], colorectal cancer (CRC) [46] and breast cancer (BrCa) [47]. Platelet count might also be an effective biomarker for monitoring disease recurrence and predicting treatment response in patients with epithelial ovarian cancer (EOC) [48], and rectal cancer [49]. Meanwhile, other platelet-associated clinical laboratory indexes including platelet to lymphocyte ratio (PLR) [50,51,52,53], platelet distribution width to platelet count ratio [54, 55], platelet to albumin ratio [56], and red cell distribution width to platelet count ratio [57] were also associated with poor progression and were shown to predict of a variety of cancers, as summarized in Table 1.
Several evidence had revealed the main molecular mechanisms of thrombocytosis (Fig. 3), including (i) tumor cells secret thrombopoietin (TPO), or interleukin-6 (IL-6) which can accelerate TPO production in the liver. TPO in turn stimulates thrombopoiesis in bone marrow [45]; (ii) TPO can stimulate differentiation, proliferation and maturation of megakaryocytes; (iii) tumor cells can accelerate platelet destruction and then induce compensatory thrombocytopenia; and (iv) malnutrition, chronic blood loss from tumor depletion, and myeloproliferative diseases can also cause thrombocytosis [58].

Mechanisms of cancer-associated thrombocytosis. Primary tumor cells secret TPO, or IL-6 which can accelerate TPO production in the liver. TPO can stimulate differentiation, proliferation and maturation of megakaryocytes in the bone marrow, as well as platelet production (TPO: thrombopoietin; IL-6: interleukin-6; MKP: megakaryocyte progenitor; MK: megakaryocyte)
Tumor cells promote production of platelet-derived substances
Moreover, cancer patients also present with increased expression levels of platelet-derived substances in the circulation, including CD40 ligand (CD40L) [59], P-selectin [60], tissue factor (TF) [61] and platelet-derived microparticles (PMPs) [62, 63]. The platelet activation markers CD40L and P-selectin play immunosuppressive effect and are used as indicators of disease progression in cancer or cancer-associated venous thromboembolism (VTE) patients [64,65,66]. It has been shown that aggressive tumors are correlated with higher levels of platelet microparticles. For example, miRNA-223 delivered by platelet-derived microparticles is significantly increased in patients with NSCLC. Tumors also induce platelet degranulation and phenotype changes in cancer patients by increasing the secretion of pro-angiogenic proteins, such as vascular endothelial growth factor (VEGF). Altogether, these studies have demonstrated cancer-activated platelets induce a procoagulant environment, providing early biomarkers for cancer screening (Table 1).
Platelets support tumor growth and metastasis
Platelets stimulate tumor angiogenesis and vascular remodeling
Platelets stimulate tumor angiogenesis through multiple mechanisms, resulting from the complex interplay between cancer cells and platelets in regulating tumor neovascularization [67]. This intercellular communication depended on the secretion of platelet α-granules, the treasure trove of the angiogenic factors in the tumor microenvironment containing VEGF and cytokines [68, 69]. In addition, the important angiogenic agents such as fibroblast growth factor (FGF), platelet-derived growth factor (PDGF) and PMPs also affect angiogenesis and indirectly enhance vessel formation [67, 69]. Thus, as an important source of angiogenesis-related factors in circulation, platelets act as “first responders” across the full spectrum of cancer progression, they and their products stimulate stroma release, promoting angiogenesis and chemotaxis [70].
In addition to regulating angiogenesis, platelets can also regulate vascular integrity, relying on the secretion of angiopoietin-1 (ANGPT1) and serotonin of α-granules, thereby promoting endothelial integrity and barrier function in primary tumors [71, 72]. While angiopoietin-2 (ANGPT2) secreted by VEGF activated endothelium could inhibit ANGPT1 competitively and destabilize vessel assembly [73]. Therefore, the stability of tumor vessel depends on the balance between the tumor and platelet-derived granules. In lymphatic vessels, platelets maintained the stability of blood-lymphatic system to support angiogenesis and tumor growth [74, 75]. Platelets might also reduce immune cells infiltration by regulating vascular integrity, reducing tissue damage by protecting tumor cells from assault of natural killer cells (NK cells) [76, 77]. Thus, platelets exhibit pro-tumorigenic functions, which directly or indirectly promote tumor growth by regulating tumor angiogenesis and vascular integrity.
Platelets support tumor invasion and metastasis
Invasion and metastasis are important features of tumorigenesis and development, and platelets also play an important role in this process. As the first cell to encounter tumor cells, it interferes with immune system surveillance to protect circulating tumor cells [78]. Upon the migration and colonization of invasive tumor cells in the blood, platelets can improve their survival and support metastatic dissemination [77]. Platelet-derived TGF-β is complexed with glycoprotein A repetitions predominant (GARP) protein to induce both NK cells and T cells anergy [79], while thrombin involved in platelet-bound GARP cleavage and the liberation of active TGF-β supports cancer immune evasion [80].
Furthermore, platelet-tumor interactions support the occurrence of epithelial-mesenchymal transformation (EMT)-like events and metastasis [81]. Platelets release EMT inducers and growth factors to shift epithelial-like phenotype to mesenchymal-like phenotype [82, 83]. Subsequently, platelet-associated cell adhesion molecules (CAMs), including integrin, P-selectin, immunoglobulin superfamily (IgSF) member glycoprotein VI (GPVI), etc. [26, 84, 85], can mediate adhesion and communication between platelets and the extracellular matrix (ECM) and among platelets to promote tumor metastasis [86]. Finally, tumor-platelet agglomerates support intravascular arrest of cancer cells via P-selectin, accelerating extravasation to distant organs [87]. In the process, platelet-secreted chemokines (like CXCL5 and CXCL7) [88], growth factors (like VEGF, PDGF, and TGF-β)[89], and PMPs-derived miRNA [90] support the proliferation, formation of pre-metastatic nitch and seeding of metastatic tumor cells. Therefore, platelets play a key role in tumor cells proliferation progression, anoikis resistance, extravasation and metastatic seeding.
To sum up, platelets are involved in the whole process of tumorigenesis and tumor development (Fig. 1). Benefit from their closed membrane structure, platelets can completely preserve the biological information of tumor sources and isolate bioactive molecules in the circulation. For these reasons, the substances carried by platelets have great potential to become tumor biomarkers (Tables 2 and 3).
Alterations and mechanisms of platelet RNA profiles in tumor
Platelet mRNA expression profiles can serve as tumor biomarkers
mRNA is the most studied type of RNA in platelets, about one-third of all human genes (~ 5000–9000 genes) mRNAs have been identified within platelets [91, 92]. Previous studies have illuminated the diagnostic value of platelet mRNA signatures as the non-invasive biomarkers for predicting tumorigenesis and monitoring tumor progression, including CRC [93], lung cancer [94], NSCLC [95], prostate cancer [96], liver cancer (hepatocellular carcinoma, HCC) [97] and etc.
Best et al. prospectively isolated, amplified, and sequenced TEP mRNA profile between healthy donors and cancer patient platelets, 5,003 differentials were identified. Using this readout, they were able to distinguish patients with localized and metastatic tumors from healthy individuals with 96% accuracy [2]. Using the R language WGCNA package, platelet RNA profiles of CRC patients and healthy donors were screened for potential biomarkers for cancer diagnostics. It was found that TIMP1 mRNA in platelets increased for tumor patients, possessing the promising diagnostic performance much higher than CEA and CA199 [93]; Besides, platelet ITGA2B levels were significantly higher in NSCLC patients than in all controls, and the combination of ITGA2B, CEA and stage could predict the overall survival [98]; A similar phenomenon was observed in a pan-cancer study, where platelets mRNA expression profiles were significantly different between tumor patients and healthy volunteers. Platelet profiles were not only suitable for cancer diagnosis, but also correctly identified the primary origins of pan-cancer. In many cases, they could accurately predict tumor gene mutation status, including MET, HER2, KRAS, EGFR or PIK3CA mutations [99]. Calverley et al. also demonstrated that they could distinguish patients with HER2 amplified, PIK3CA mutant or triple-negative BrCa (TNBC) and NSCLC patients with MET overexpression, although the low levels of these mutant biomarkers needed to be considered [99]. Our previous study also demonstrated significant changes in platelet mRNA expression profiles in lung cancer patients [100]. A total of 1306 mRNAs with the differential expression were identified, among which MAX, MTURN, UQCRH and HLA-B were significantly upregulated and correlated with ‘‘favorable’’ first chemotherapy response, thus providing a noninvasive marker to predict first chemotherapy response.
Splicing is the major regulatory mechanism for TEP mRNA expression
Although mature platelets are anucleate, they still retain endogenous pre-mRNAs inherited from the transcription of nuclear DNA in the megakaryocyte as well exploit functional spliceosome [101]. Once activated by external signals, such as activation of platelet surface receptors and lipopolysaccharide-mediated platelet activation, these transcripts can be specifically spliced into mature mRNA and translated into thousands of different proteins [102]. RNA splicing is closely related to changes in platelet mRNA profiles, and analysis demonstrated that pre-mRNA splicing might occur during platelet activation [103]. For example, interleukin-1β (IL-1β) was spliced into mature mRNA transcripts, resulting in the synthesis of IL-1b proteins in response to cellular activation in quiescent platelets [101, 104, 105].
Aberrant RNA splicing is an underlying highly conserved process, occurring in > 95% of human multi-exon genes [106]. A Pan-Cancer study have found an average of 20% more alternative splicing in tumors than in corresponding healthy tissues [107]. Platelets may also undergo queue-specific splice events in response to signals released by cancer cells and tumor microenvironment [102]. The specific splice events can provide platelets with a highly dynamic mRNA repertoire in patients with different types and organs of tumors, with potential applicability to cancer diagnosis and tumor origin tracking [95, 99]. Previous research detected the differential expression of spliced RNAs in NSCLC patients based on the intron-spanning read count analysis. They identified 1,625 spliced platelet genes with significantly different spliced levels (698 genes with enhanced splicing in platelets of NSCLC patients and 927 genes with decreased splicing in platelets of NSCLC patients). The most significantly enriched spliced RNAs in TEPs included CFL1, ACOT7, and ARPC1B, whereas DDX5, RPS5, and EEF1B2 were decreased [95]. Therefore, a large number of changes in platelet splicing behavior during platelet activation are undoubtedly one of the main reasons for the changes in platelet mRNA expression profiles (Fig. 4).

Platelets exploit a functional spliceosome for pre-mRNA splicing. Megakaryocytes sort distinctive RNA molecules into proplatelets during thrombopoiesis. Pre-mRNAs contain exons and introns and are processed by U snRNPs that make up the spliceosome. Platelet spliceosomes alternatively excise introns from pre-mRNA, yielding a mature message that is translated into protein
TEP snRNAs as novel biomarker in cancer detection
snRNPs dominate RNA splicing
Chemical reactions of pre-mRNA splicing in platelets occur only after the pre-mRNA assembles into the functional spliceosome, a multi-component complex termed as snRNPs composed of U1, U2, U4, U5, U6 snRNAs and their associated protein components [10, 11], including a protein-only NineTeen Complex (NTC) and a number of accessory proteins [108, 109]. It has been shown that platelets contain many spliceosome-associated proteins, including U1 70 K, U2AF, SRm160, SMN, and SF2/ASF [7], as well as snRNAs, which direct the accurate removal of intronic sequences from pre-mRNAs.
During spliceosome assembly, snRNAs and splicing factors recognize and interact with the pre-mRNA consensus sequences, facilitating and specifying the transesterification reactions [110]. Their main process in the spliceosome complex is that U1 and U2 snRNPs are responsible for recognizing the 5′ splice site and branchpoint upstream of the 3′ splice site, and U4/U6.U5 tri-snRNP is added to the spliceosome before rearrangements, guiding U6 snRNP to catalyze the actual splicing reaction [13, 111]. In addition to U1, U2, U4, U5, and U6 snRNPs (major), other minor spliceosome snRNP species (U11, U12, U4atac, U5atac and U6atac) are also involved in splicing a minor class of introns [112, 113]. Eventually, introns are removed, and protein-coding segments known as exons are spliced together to form mRNAs [114, 115]. It was previously thought that spliceosome components were only present in nucleated cells [116], but later it was reported that anucleate platelets also exploit the functional spliceosome inherited from megakaryocytes during thrombopoiesis [101]. More Importantly, snRNAs are not merely the basal factors ubiquitously expressed in all cells [12], whereas they are extremely variable across a wide range of biological conditions [13].
Alternation of snRNAs regulate alternative splicing in cancer
Recently, snRNPs have been shown to act as regulatory molecules to mediate cancer processes through alternative splicing [117, 118]. It can directly or indirectly affect too many molecular targets, thereby regulating cis-acting elements, transacting factors, or pre-mRNA transcription at multiple levels [119]. In particular, endogenous variation at snRNA levels leads to widespread differences in alternative splicing. Studies have shown that snRNA dysregulation shapes the transcriptome of breast cancer [13], exhibiting subtype-specific dependence on the abundance of different snRNAs [120, 121]. For example, the HER2 subtype shows high levels of U1 and U5A, while triple-negative samples have high abundance of U6 or relatively low levels of U2 and U5A [122].
SnRNAs can also be subject to somatic mutations in addition to aberrant expression, which can alter the normal splicing process to drive heredity, dysplasia, and even tumorigenesis and cancer progression [123, 124]. For example, aberrant U1 snRNA (A > C somatic mutation at the third base of U1) has been reported in several tumor types, generating novel splice junctions and altering the splicing pattern of multiple genes.
Alternation of platelet snRNAs
Multiple hypotheses exist regarding the source and mechanism of platelet snRNAs alterations. One hypothesis supposes RNA expression patterns are fluid throughout megakaryocyte development and platelet biogenesis [125, 126]. Alterations of platelet snRNAs are caused by RNA differential sorting mechanism of megakaryocytes [127]. In addition, an alternative source mechanism has recently been discussed, namely the ability of extracellular vehicles (EVs) to transmit snRNAs horizontally [9]. Circulating platelets can capture and store tumor-derived EVs from the periphery, and then obtain characteristic biological information, which is one of the main mechanisms of TEP.
It has recently been shown that megakaryocytes selectively sort RNAs into platelets rather than randomly, allowing only a fraction of RNAs transferred into platelets. This observation is supported by a recent study describing how megakaryocytes preferentially sorted matrix metalloproteases (MMPs) and their tissue inhibitors into platelets [127]. Nevertheless, the sorting mechanisms appear largely unknown [128]. Few studies have expounded whether changes in the megakaryocyte environment would alter the types and amounts of RNA sorting to platelets [129].
EVs also have the ability to transmit information to platelets horizontally [99]. EVs are membrane-separated subcellular particles containing a variety of biologically active molecules. They are the main messengers of local and systemic intercellular biological information exchange [130], and contain nearly all types of non-protein-coding RNAs (ncRNAs), which can be transferred horizontally between cells regulating gene expression and the malignant phenotype in recipient cells, [131]. The results of deep RNA sequencing showed that the proportion of snRNAs was 25%, accounting for the majority of all short ncRNAs in cells, among which 11% in microvesicles (MVs), and 20% in exosomes [132]. While another study confirmed that the expression level of snRNA RNU6-1 was significantly increased in serum EVs of neuroblastoma patients [133]. Our previous research also reported that TEP U1, U2 and U5 levels were closely correlated between platelet and paired exosomes, indicating that snRNAs might be released from tumors to educate platelets through EVs [134].
Alterations of snRNAs as cancer biomarker
As shown in Table 4, alterations of snRNAs have been reported in multiple tumors. It was reported in the 1102-case research that three differential snRNAs including RNU1-106 P, RNU6-850 P, and RNU6-529 P were found in pan-adenocarcinomas of the esophagus, stomach, colon, and rectum digestive tract, with potential as the biomarkers for diagnosis and progression monitoring for cancer [135]. Moreover, U2 is one of the most highly-expressed in blood and widely-studied snRNAs as a potential tumor marker [136]. Fragments derived from U2 snRNA (RNU2-1f) were differentially expressed in a variety of tumors, with the upregulation not only in serum [137,138,139,140,141] but also in cerebrospinal fluid [142], serving as the potential diagnostic biomarker. It also acts as the prognostic factor. Its relatively high expression of serum RNU2-1f was closely related to shorter median survival in lung cancer patients [137] and a high risk of recurrence and poor prognosis in ovarian cancer [139] (Table 4).
In the previous experiment, we validated whether TEP snRNAs served as the potential biomarkers for lung cancer [134]. TEP U1, U2 and U5 levels were significantly decreased in lung cancer patients, possessing the favorable diagnostic efficiency, especially in early lung cancer. Moreover, their downregulation was correlated with lung cancer progression. It was coincided with previous reports, 99% of differential mRNAs in TEP of untreated lung cancer patients were down-regulated [143]. This might also explain the accumulation of a large number of immature reticulated platelets in the blood of NSCLC patients and the down-regulation of splicing function blocked the maturation of reticulated platelets [95].
Conclusion and perspective
The bidirectional tumor-platelet interactions are reciprocal and complicated, during which the platelets are educated by tumor and derived bio-substance, and empowered with the potential to identify surrogate biomarker signatures to detect cancer. Multiple studies have shown that platelet-based biomarkers (e.g., count, volume, RNA profile and protein profile) are incorporated into liquid biopsy platforms [144]. As the liquid biopsy tool, platelets are easily isolated and counted and are the second most abundant cell in circulation, thus making them more attractive for clinical applications [2]. Moreover, platelets occupies the short life span (average of 7 days), and more importantly, splicing activity and rapid protein translation, thereby the contents in TEPs are dynamic and transient in response to external stimuli, providing the opportunity to potentially serve as a promising diagnostic, prognostic, and therapeutic tool that enables high specificity and sensitivity in the search for new ways to fight against malignancies [145].
The unique benefits of TEP for cancer detection are exciting, nevertheless, some limitations should be taken into consideration. It has been reported that the same RNA plays different roles between cells and platelets, indicating different splicing mechanisms in platelets from those in cells [119]. It has been observed that cancer cells disrupt normal alternative splicing events to generate specialized splicing isoforms that affect cell function and control cell proliferation and tumorigenesis [146,147,148]. Although TEPs as a novel biosource for cancer diagnostics are widely recognized, it is generally uninformed about the mechanisms how conformational and compositional changes within the spliceosome determine splicing outcomes [109], which urgently needs further investigation to enable extended and more optimal diagnostics. Besides, there is still a large gap between biomarker discovery and clinical validation and implementation. The simplified, low-cost and standardized methodologies must be developed. For example, the most commonly used method of platelet isolation is low-speed centrifugation, but the protocols quite differ from different researches and laboratories [149]. Therefore, consensus on methods for TEP research of normalization, sample collection, and processing is essential and imperative. Another critical point for the TEP clinical implementation would be to perform clinical utility studies. A dedicated, well-powered, blinded, and population-targeted prospective clinical trial based on TEP platform should be further pursued as other types of liquid biopsies to ensure the clinical value of platelet-related biomarkers including RNA splicing signatures [150]. Collectively, we believe that TEP RNA repertoire and RNA processing machineries including snRNAs will be widely used in cancer diagnosis, treatment and prognosis monitoring in future, bringing great progress to the cancer diagnostics and treatment and warrant further research.
Availability of data and materials
The data supporting the conclusions of this article will be made available by the authors, without undue reservation.
References
Palacios-Acedo AL, Mege D, Crescence L, Dignat-George F, Dubois C, Panicot-Dubois L, Platelets. Thrombo-Inflammation, and Cancer: collaborating with the enemy. Front Immunol. 2019;10:1805.
Haemmerle M, Stone RL, Menter DG, Afshar-Kharghan V, Sood AK. The platelet lifeline to Cancer: Challenges and Opportunities. Cancer Cell. 2018;33(6):965–83.
Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–57.
Goubran HA, Burnouf T, Stakiw J, Seghatchian J. Platelet microparticle: a sensitive physiological “fine tuning” balancing factor in health and disease. Transfus Apher Sci. 2015;52(1):12–8.
Plantureux L, Crescence L, Dignat-George F, Panicot-Dubois L, Dubois C. Effects of platelets on cancer progression. Thromb Res. 2018;164(Suppl 1):40–S7.
Plantureux L, Mege D, Crescence L, Dignat-George F, Dubois C, Panicot-Dubois L. Impacts of Cancer on Platelet Production, Activation and Education and Mechanisms of Cancer-Associated Thrombosis. Cancers (Basel). 2018;10(11).
Best MG, Vancura A, Wurdinger T. Platelet RNA as a circulating biomarker trove for cancer diagnostics. J Thromb Haemost. 2017;15(7):1295–306.
Angenieux C, Maitre B, Eckly A, Lanza F, Gachet C, de la Salle H. Time-Dependent Decay of mRNA and ribosomal RNA during platelet aging and its correlation with translation activity. PLoS ONE. 2016;11(1):e0148064.
Nilsson RJ, Balaj L, Hulleman E, van Rijn S, Pegtel DM, Walraven M, et al. Blood platelets contain tumor-derived RNA biomarkers. Blood. 2011;118(13):3680–3.
Yamazaki N, Kanazawa K, Kimura M, Ike H, Shinomiya M, Tanaka S, et al. Use of modified U1 small nuclear RNA for rescue from exon 7 skipping caused by 5’-splice site mutation of human cathepsin A gene. Gene. 2018;677:41–8.
Qin M, Wei G, Sun X. Circ-UBR5: an exonic circular RNA and novel small nuclear RNA involved in RNA splicing. Biochem Biophys Res Commun. 2018;503(2):1027–34.
Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136(4):701–18.
Dvinge H, Guenthoer J, Porter PL, Bradley RK. RNA components of the spliceosome regulate tissue- and cancer-specific alternative splicing. Genome Res. 2019;29(10):1591–604.
El Marabti E, Younis I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front Mol Biosci. 2018;5:80.
Wang Y, Zhang H, Li H, Xiong J, Wang J, Huang Y. Application of tumor-educated platelets as new fluid biopsy markers in various tumors. Clin Transl Oncol. 2023;25(1):114–25.
Morris K, Schnoor B, Papa AL. Platelet cancer cell interplay as a new therapeutic target. Biochim Biophys Acta Rev Cancer. 2022;1877(5):188770.
Wang L, Wang X, Guo E, Mao X, Miao S. Emerging roles of platelets in cancer biology and their potential as therapeutic targets. Front Oncol. 2022;12:939089.
Xu XR, Yousef GM, Ni H. Cancer and platelet crosstalk: opportunities and challenges for aspirin and other antiplatelet agents. Blood. 2018;131(16):1777–89.
Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22.
Lonsdorf AS, Kramer BF, Fahrleitner M, Schonberger T, Gnerlich S, Ring S, et al. Engagement of alphaIIbbeta3 (GPIIb/IIIa) with alphanubeta3 integrin mediates interaction of melanoma cells with platelets: a connection to hematogenous metastasis. J Biol Chem. 2012;287(3):2168–78.
Goh CY, Patmore S, Smolenski A, Howard J, Evans S, O’Sullivan J, et al. The role of von Willebrand factor in breast cancer metastasis. Transl Oncol. 2021;14(4):101033.
Qi Y, Chen W, Liang X, Xu K, Gu X, Wu F, et al. Novel antibodies against GPIbalpha inhibit pulmonary metastasis by affecting vWF-GPIbalpha interaction. J Hematol Oncol. 2018;11(1):117.
Lei X, Reheman A, Hou Y, Zhou H, Wang Y, Marshall AH, et al. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thromb Haemost. 2014;111(2):279–89.
Karakas D, Xu M, Ni H. GPIbalpha is the driving force of hepatic thrombopoietin generation. Res Pract Thromb Haemost. 2021;5(4):e12506.
Mammadova-Bach E, Ollivier V, Loyau S, Schaff M, Dumont B, Favier R, et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood. 2015;126(5):683–91.
Mammadova-Bach E, Gil-Pulido J, Sarukhanyan E, Burkard P, Shityakov S, Schonhart C, et al. Platelet glycoprotein VI promotes metastasis through interaction with cancer cell-derived galectin-3. Blood. 2020;135(14):1146–60.
Mammadova-Bach E, Zigrino P, Brucker C, Bourdon C, Freund M, De Arcangelis A, et al. Platelet integrin alpha6beta1 controls lung metastasis through direct binding to cancer cell-derived ADAM9. JCI Insight. 2016;1(14):e88245.
Carpinteiro A, Becker KA, Japtok L, Hessler G, Keitsch S, Pozgajova M, et al. Regulation of hematogenous tumor metastasis by acid sphingomyelinase. EMBO Mol Med. 2015;7(6):714–34.
Carpinteiro A, Beckmann N, Seitz A, Hessler G, Wilker B, Soddemann M, et al. Role of Acid Sphingomyelinase-Induced Signaling in Melanoma cells for Hematogenous Tumor Metastasis. Cell Physiol Biochem. 2016;38(1):1–14.
Shirai T, Inoue O, Tamura S, Tsukiji N, Sasaki T, Endo H, et al. C-type lectin-like receptor 2 promotes hematogenous tumor metastasis and prothrombotic state in tumor-bearing mice. J Thromb Haemost. 2017;15(3):513–25.
Astarita JL, Acton SE, Turley SJ. Podoplanin: emerging functions in development, the immune system, and cancer. Front Immunol. 2012;3:283.
Riedl J, Preusser M, Nazari PM, Posch F, Panzer S, Marosi C, et al. Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood. 2017;129(13):1831–9.
Chen C, He Z, Sai P, Faridi A, Aziz A, Kalavar M, et al. Inhibition of human CD24 binding to platelet-bound P-selectin by monoclonal antibody. Proc West Pharmacol Soc. 2004;47:28–9.
Kim YJ, Borsig L, Varki NM, Varki A. P-selectin deficiency attenuates tumor growth and metastasis. Proc Natl Acad Sci U S A. 1998;95(16):9325–30.
Garcia J, Callewaert N, Borsig L. P-selectin mediates metastatic progression through binding to sulfatides on tumor cells. Glycobiology. 2007;17(2):185–96.
Schwarz S, Schlesinger M, Bendas G. Detection of Tumor Cell-Induced platelet aggregation and granule secretion. Methods Mol Biol. 2021;2294:181–95.
Zara M, Canobbio I, Visconte C, Canino J, Torti M, Guidetti GF. Molecular mechanisms of platelet activation and aggregation induced by breast cancer cells. Cell Signal. 2018;48:45–53.
Menter DG, Hatfield JS, Harkins C, Sloane BF, Taylor JD, Crissman JD, et al. Tumor cell-platelet interactions in vitro and their relationship to in vivo arrest of hematogenously circulating tumor cells. Clin Exp Metastasis. 1987;5(1):65–78.
Italiano JE Jr, Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111(3):1227–33.
Mitrugno A, Williams D, Kerrigan SW, Moran N. A novel and essential role for FcgammaRIIa in cancer cell-induced platelet activation. Blood. 2014;123(2):249–60.
Ikeda M, Furukawa H, Imamura H, Shimizu J, Ishida H, Masutani S, et al. Poor prognosis associated with thrombocytosis in patients with gastric cancer. Ann Surg Oncol. 2002;9(3):287–91.
Bailey SE, Ukoumunne OC, Shephard E, Hamilton W. How useful is thrombocytosis in predicting an underlying cancer in primary care? A systematic review. Fam Pract. 2017;34(1):4–10.
Abdel-Razeq H, Mansour A, Saadeh SS, Abu-Nasser M, Makoseh M, Salam M, et al. The application of current proposed venous thromboembolism risk Assessment Model for ambulatory patients with Cancer. Clin Appl Thromb Hemost. 2018;24(3):429–33.
Pedersen LM, Milman N. Prognostic significance of thrombocytosis in patients with primary lung cancer. Eur Respir J. 1996;9(9):1826–30.
Stone RL, Nick AM, McNeish IA, Balkwill F, Han HD, Bottsford-Miller J, et al. Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med. 2012;366(7):610–8.
Sasaki K, Kawai K, Tsuno NH, Sunami E, Kitayama J. Impact of preoperative thrombocytosis on the survival of patients with primary colorectal cancer. World J Surg. 2012;36(1):192–200.
Taucher S, Salat A, Gnant M, Kwasny W, Mlineritsch B, Menzel RC, et al. Impact of pretreatment thrombocytosis on survival in primary breast cancer. Thromb Haemost. 2003;89(6):1098–106.
Hu Q, Hada A, Han L. Platelet count as a biomarker for monitoring treatment response and disease recurrence in recurrent epithelial ovarian cancer. J Ovarian Res. 2020;13(1):78.
Toiyama Y, Inoue Y, Kawamura M, Kawamoto A, Okugawa Y, Hiro J, et al. Elevated platelet count as predictor of recurrence in rectal cancer patients undergoing preoperative chemoradiotherapy followed by surgery. Int Surg. 2015;100(2):199–207.
Gong Z, Xin R, Li L, Lv L, Wu X. Platelet-to-lymphocyte ratio associated with the clinicopathological features and prognostic value of breast cancer: A meta-analysis. Int J Biol Markers. 2022:3936155221118098.
Pan Y, Si H, Deng G, Chen S, Zhang N, Zhou Q, et al. A Composite Biomarker of Derived Neutrophil-Lymphocyte ratio and platelet-lymphocyte ratio correlates with outcomes in Advanced gastric Cancer patients treated with Anti-PD-1 antibodies. Front Oncol. 2021;11:798415.
Matsuda A, Yamada T, Matsumoto S, Shinji S, Ohta R, Sonoda H, et al. Prognostic role of the platelet-to-lymphocyte ratio for patients with metastatic colorectal Cancer treated with Aflibercept. In Vivo. 2020;34(5):2667–73.
Mandaliya H, Jones M, Oldmeadow C, Nordman II. Prognostic biomarkers in stage IV non-small cell lung cancer (NSCLC): neutrophil to lymphocyte ratio (NLR), lymphocyte to monocyte ratio (LMR), platelet to lymphocyte ratio (PLR) and advanced lung cancer inflammation index (ALI). Transl Lung Cancer Res. 2019;8(6):886–94.
Shen Y, Xu H, Guan Z, Lv M, Qian T, Wu Y. Effect of rho GTPase activating protein 9 combined with preoperative ratio of platelet distribution width to platelet count on prognosis of patients with serous ovarian cancer. Transl Cancer Res. 2021;10(10):4440–53.
Takeuchi H, Abe M, Takumi Y, Hashimoto T, Kobayashi R, Osoegawa A, et al. The prognostic impact of the platelet distribution width-to-platelet count ratio in patients with breast cancer. PLoS ONE. 2017;12(12):e0189166.
Guo M, Sun T, Zhao Z, Ming L. Preoperative platelet to albumin ratio predicts outcome of patients with non-small-cell Lung Cancer. Ann Thorac Cardiovasc Surg. 2021;27(2):84–90.
Takeuchi H, Abe M, Takumi Y, Hashimoto T, Miyawaki M, Okamoto T, et al. Elevated red cell distribution width to platelet count ratio predicts poor prognosis in patients with breast cancer. Sci Rep. 2019;9(1):3033.
Kaser A, Brandacher G, Steurer W, Kaser S, Offner FA, Zoller H, et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood. 2001;98(9):2720–5.
Riedl J, Hell L, Kaider A, Koder S, Marosi C, Zielinski C, et al. Association of platelet activation markers with cancer-associated venous thromboembolism. Platelets. 2016;27(1):80–5.
Ay C, Pabinger I. Predictive potential of haemostatic biomarkers for venous thromboembolism in cancer patients. Thromb Res. 2012;129(Suppl 1):6–9.
Freyssinet JM, Toti F. Formation of procoagulant microparticles and properties. Thromb Res. 2010;125(Suppl 1):46–8.
Rank A, Liebhardt S, Zwirner J, Burges A, Nieuwland R, Toth B. Circulating microparticles in patients with benign and malignant ovarian tumors. Anticancer Res. 2012;32(5):2009–14.
Reddel CJ, Tan CW, Chen VM. Thrombin Generation and Cancer: Contributors and Consequences. Cancers (Basel). 2019;11(1).
Huang J, Jochems C, Talaie T, Anderson A, Jales A, Tsang KY, et al. Elevated serum soluble CD40 ligand in cancer patients may play an immunosuppressive role. Blood. 2012;120(15):3030–8.
Herold Z, Herold M, Herczeg G, Fodor A, Szasz AM, Dank M, et al. High plasma CD40 ligand level is associated with more advanced stages and worse prognosis in colorectal cancer. World J Clin Cases. 2022;10(13):4084–96.
Dymicka-Piekarska V, Korniluk A, Gryko M, Siergiejko E, Kemona H. Potential role of soluble CD40 ligand as inflammatory biomarker in colorectal cancer patients. Int J Biol Markers. 2014;29(3):e261–7.
Eelen G, Treps L, Li X, Carmeliet P. Basic and Therapeutic Aspects of Angiogenesis updated. Circ Res. 2020;127(2):310–29.
Klement GL, Yip TT, Cassiola F, Kikuchi L, Cervi D, Podust V, et al. Platelets actively sequester angiogenesis regulators. Blood. 2009;113(12):2835–42.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307.
Menter DG, Kopetz S, Hawk E, Sood AK, Loree JM, Gresele P, et al. Platelet “first responders” in wound response, cancer, and metastasis. Cancer Metastasis Rev. 2017;36(2):199–213.
Li JJ, Huang YQ, Basch R, Karpatkin S. Thrombin induces the release of angiopoietin-1 from platelets. Thromb Haemost. 2001;85(2):204–6.
Ho-Tin-Noe B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008;68(16):6851–8.
Qin S, Yi M, Jiao D, Li A, Wu K. Distinct roles of VEGFA and ANGPT2 in lung adenocarcinoma and squamous cell carcinoma. J Cancer. 2020;11(1):153–67.
Bertozzi CC, Schmaier AA, Mericko P, Hess PR, Zou Z, Chen M, et al. Platelets regulate lymphatic vascular development through CLEC-2-SLP-76 signaling. Blood. 2010;116(4):661–70.
Haining EJ, Lowe KL, Wichaiyo S, Kataru RP, Nagy Z, Kavanagh DP, et al. Lymphatic blood filling in CLEC-2-deficient mouse models. Platelets. 2021;32(3):352–67.
Nieswandt B, Hafner M, Echtenacher B, Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999;59(6):1295–300.
Schlesinger M. Role of platelets and platelet receptors in cancer metastasis. J Hematol Oncol. 2018;11(1):125.
Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012;2(12):1091–9.
Rachidi S, Metelli A, Riesenberg B, Wu BX, Nelson MH, Wallace C et al. Platelets subvert T cell immunity against cancer via GARP-TGFbeta axis. Sci Immunol. 2017;2(11).
Metelli A, Wu BX, Riesenberg B, Guglietta S, Huck JD, Mills C et al. Thrombin contributes to cancer immune evasion via proteolysis of platelet-bound GARP to activate LTGF-beta. Sci Transl Med. 2020;12(525).
Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2020;21(6):341–52.
Xiong G, Chen J, Zhang G, Wang S, Kawasaki K, Zhu J, et al. Hsp47 promotes cancer metastasis by enhancing collagen-dependent cancer cell-platelet interaction. Proc Natl Acad Sci U S A. 2020;117(7):3748–58.
McCarty OJ, Mousa SA, Bray PF, Konstantopoulos K. Immobilized platelets support human colon carcinoma cell tethering, rolling, and firm adhesion under dynamic flow conditions. Blood. 2000;96(5):1789–97.
Harjunpaa H, Llort Asens M, Guenther C, Fagerholm SC. Cell adhesion molecules and their roles and regulation in the Immune and Tumor Microenvironment. Front Immunol. 2019;10:1078.
Kadry YA, Calderwood DA. Chapter 22: structural and signaling functions of integrins. Biochim Biophys Acta Biomembr. 2020;1862(5):183206.
Liu Y, Zhang Y, Ding Y, Zhuang R. Platelet-mediated tumor metastasis mechanism and the role of cell adhesion molecules. Crit Rev Oncol Hematol. 2021;167:103502.
Shao B, Wahrenbrock MG, Yao L, David T, Coughlin SR, Xia L, et al. Carcinoma mucins trigger reciprocal activation of platelets and neutrophils in a murine model of Trousseau syndrome. Blood. 2011;118(15):4015–23.
Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. Proc Natl Acad Sci U S A. 2014;111(30):E3053–61.
Heldin CH, Westermark B, Wasteson A. Platelet-derived growth factor. Isolation by a large-scale procedure and analysis of subunit composition. Biochem J. 1981;193(3):907–13.
Gidlof O, van der Brug M, Ohman J, Gilje P, Olde B, Wahlestedt C, et al. Platelets activated during myocardial infarction release functional miRNA, which can be taken up by endothelial cells and regulate ICAM1 expression. Blood. 2013;121(19):3908.
Gnatenko DV, Dunn JJ, Schwedes J, Bahou WF. Transcript profiling of human platelets using microarray and serial analysis of gene expression (SAGE). Methods Mol Biol. 2009;496:245–72.
Bugert P, Dugrillon A, Gunaydin A, Eichler H, Kluter H. Messenger RNA profiling of human platelets by microarray hybridization. Thromb Haemost. 2003;90(4):738–48.
Yang L, Jiang Q, Li DZ, Zhou X, Yu DS, Zhong J. TIMP1 mRNA in tumor-educated platelets is diagnostic biomarker for colorectal cancer. Aging. 2019;11(20):8998–9012.
Liu L, Lin F, Ma X, Chen Z, Yu J. Tumor-educated platelet as liquid biopsy in lung cancer patients. Crit Rev Oncol Hematol. 2020;146:102863.
Best MG, Sol N, In ‘t Veld S, Vancura A, Muller M, Niemeijer AN, et al. Swarm Intelligence-Enhanced detection of Non-Small-Cell Lung Cancer using tumor-educated platelets. Cancer Cell. 2017;32(2):238–52e9.
Tjon-Kon-Fat LA, Lundholm M, Schroder M, Wurdinger T, Thellenberg-Karlsson C, Widmark A, et al. Platelets harbor prostate cancer biomarkers and the ability to predict therapeutic response to abiraterone in castration resistant patients. Prostate. 2018;78(1):48–53.
Best MG, Wurdinger T. Tumor-educated platelets for the earlier detection of hepatocellular carcinoma. Clin Res Hepatol Gastroenterol. 2020;44(6):794–5.
Xing S, Zeng T, Xue N, He Y, Lai YZ, Li HL, et al. Development and Validation of Tumor-educated blood platelets integrin alpha 2b (ITGA2B) RNA for diagnosis and prognosis of non-small-cell Lung Cancer through RNA-seq. Int J Biol Sci. 2019;15(9):1977–92.
Best MG, Sol N, Kooi I, Tannous J, Westerman BA, Rustenburg F, et al. RNA-Seq of Tumor-Educated platelets enables blood-based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell. 2015;28(5):666–76.
Liu L, Song X, Li X, Xue L, Ding S, Niu L, et al. A three-platelet mRNA set: MAX, MTURN and HLA-B as biomarker for lung cancer. J Cancer Res Clin Oncol. 2019;145(11):2713–23.
Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005;122(3):379–91.
Best MG, Wesseling P, Wurdinger T. Tumor-educated platelets as a noninvasive biomarker source for Cancer Detection and Progression Monitoring. Cancer Res. 2018;78(13):3407–12.
Brogren H, Karlsson L, Andersson M, Wang L, Erlinge D, Jern S. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood. 2004;104(13):3943–8.
Weyrich AS, Dixon DA, Pabla R, Elstad MR, McIntyre TM, Prescott SM, et al. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci U S A. 1998;95(10):5556–61.
Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001;154(3):485–90.
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–6.
Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 patients. Cancer Cell. 2018;34(2):211–24. e6.
Karijolich J, Yu YT. Spliceosomal snRNA modifications and their function. RNA Biol. 2010;7(2):192–204.
Didychuk AL, Butcher SE, Brow DA. The life of U6 small nuclear RNA, from cradle to grave. RNA. 2018;24(4):437–60.
Hoskins AA, Moore MJ. The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem Sci. 2012;37(5):179–88.
Fica SM, Tuttle N, Novak T, Li NS, Lu J, Koodathingal P, et al. RNA catalyses nuclear pre-mRNA splicing. Nature. 2013;503(7475):229–34.
Morais P, Adachi H, Yu YT. Spliceosomal snRNA Epitranscriptomics Front Genet. 2021;12:652129.
Gadgil A, Raczynska KD. U7 snRNA: a tool for gene therapy. J Gene Med. 2021;23(4):e3321.
Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15(2):108–21.
Wilkinson ME, Charenton C, Nagai K. RNA splicing by the spliceosome. Annu Rev Biochem. 2020;89:359–88.
Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002;416(6880):499–506.
Cheng Z, Sun Y, Niu X, Shang Y, Ruan J, Chen Z, et al. Gene expression profiling reveals U1 snRNA regulates cancer gene expression. Oncotarget. 2017;8(68):112867–74.
Oh JM, Venters CC, Di C, Pinto AM, Wan L, Younis I, et al. U1 snRNP regulates cancer cell migration and invasion in vitro. Nat Commun. 2020;11(1):1.
Liu Y, Liu X, Lin C, Jia X, Zhu H, Song J, et al. Noncoding RNAs regulate alternative splicing in Cancer. J Exp Clin Cancer Res. 2021;40(1):11.
Chan S, Sridhar P, Kirchner R, Lock YJ, Herbert Z, Buonamici S, et al. Basal-A triple-negative breast Cancer cells selectively rely on RNA splicing for Survival. Mol Cancer Ther. 2017;16(12):2849–61.
Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525(7569):384–8.
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–74.
Suzuki H, Kumar SA, Shuai S, Diaz-Navarro A, Gutierrez-Fernandez A, De Antonellis P, et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature. 2019;574(7780):707–11.
Jankowska A, Gunderson SI, Andrusiewicz M, Burczynska B, Szczerba A, Jarmolowski A, et al. Reduction of human chorionic gonadotropin beta subunit expression by modified U1 snRNA caused apoptosis in cervical cancer cells. Mol Cancer. 2008;7:26.
Opalinska JB, Bersenev A, Zhang Z, Schmaier AA, Choi J, Yao Y, et al. MicroRNA expression in maturing murine megakaryocytes. Blood. 2010;116(23):e128–38.
Bluteau O, Langlois T, Rivera-Munoz P, Favale F, Rameau P, Meurice G, et al. Developmental changes in human megakaryopoiesis. J Thromb Haemost. 2013;11(9):1730–41.
Cecchetti L, Tolley ND, Michetti N, Bury L, Weyrich AS, Gresele P. Megakaryocytes differentially sort mRNAs for matrix metalloproteinases and their inhibitors into platelets: a mechanism for regulating synthetic events. Blood. 2011;118(7):1903–11.
Risitano A, Beaulieu LM, Vitseva O, Freedman JE. Platelets and platelet-like particles mediate intercellular RNA transfer. Blood. 2012;119(26):6288–95.
Rondina MT, Weyrich AS. Regulation of the genetic code in megakaryocytes and platelets. J Thromb Haemost. 2015;13(Suppl 1):26–32.
Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in Cancer: cell-to-cell mediators of Metastasis. Cancer Cell. 2016;30(6):836–48.
Anfossi S, Babayan A, Pantel K, Calin GA. Clinical utility of circulating non-coding RNAs - an update. Nat Rev Clin Oncol. 2018;15(9):541–63.
Wei Z, Batagov AO, Schinelli S, Wang J, Wang Y, El Fatimy R, et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat Commun. 2017;8(1):1145.
Manterola L, Guruceaga E, Gallego Perez-Larraya J, Gonzalez-Huarriz M, Jauregui P, Tejada S, et al. A small noncoding RNA signature found in exosomes of GBM patient serum as a diagnostic tool. Neuro Oncol. 2014;16(4):520–7.
Dong X, Ding S, Yu M, Niu L, Xue L, Zhao Y, et al. Small Nuclear RNAs (U1, U2, U5) in tumor-educated platelets are downregulated and act as promising biomarkers in Lung Cancer. Front Oncol. 2020;10:1627.
Qin XG, Zeng JH, Lin P, Mo WJ, Li Q, Feng ZB, et al. Prognostic value of small nuclear RNAs (snRNAs) for digestive tract pan- adenocarcinomas identified by RNA sequencing data. Pathol Res Pract. 2019;215(3):414–26.
Mazieres J, Catherinne C, Delfour O, Gouin S, Rouquette I, Delisle MB, et al. Alternative processing of the U2 small nuclear RNA produces a 19-22nt fragment with relevance for the detection of non-small cell lung cancer in human serum. PLoS ONE. 2013;8(3):e60134.
Kohler J, Schuler M, Gauler TC, Nopel-Dunnebacke S, Ahrens M, Hoffmann AC, et al. Circulating U2 small nuclear RNA fragments as a diagnostic and prognostic biomarker in lung cancer patients. J Cancer Res Clin Oncol. 2016;142(4):795–805.
Baraniskin A, Nopel-Dunnebacke S, Ahrens M, Jensen SG, Zollner H, Maghnouj A, et al. Circulating U2 small nuclear RNA fragments as a novel diagnostic biomarker for pancreatic and colorectal adenocarcinoma. Int J Cancer. 2013;132(2):E48–57.
Kuhlmann JD, Baraniskin A, Hahn SA, Mosel F, Bredemeier M, Wimberger P, et al. Circulating U2 small nuclear RNA fragments as a novel diagnostic tool for patients with epithelial ovarian cancer. Clin Chem. 2014;60(1):206–13.
Baraniskin A, Nopel-Dunnebacke S, Schumacher B, Gerges C, Bracht T, Sitek B, et al. Analysis of U2 small nuclear RNA fragments in the bile differentiates cholangiocarcinoma from primary sclerosing cholangitis and other benign biliary disorders. Dig Dis Sci. 2014;59(7):1436–41.
Kuhlmann JD, Wimberger P, Wilsch K, Fluck M, Suter L, Brunner G. Increased level of circulating U2 small nuclear RNA fragments indicates metastasis in melanoma patients. Clin Chem Lab Med. 2015;53(4):605–11.
Baraniskin A, Zaslavska E, Nopel-Dunnebacke S, Ahle G, Seidel S, Schlegel U, et al. Circulating U2 small nuclear RNA fragments as a novel diagnostic biomarker for primary central nervous system lymphoma. Neuro Oncol. 2016;18(3):361–7.
Calverley DC, Phang TL, Choudhury QG, Gao B, Oton AB, Weyant MJ, et al. Significant downregulation of platelet gene expression in metastatic lung cancer. Clin Transl Sci. 2010;3(5):227–32.
Chen M, Hou L, Hu L, Tan C, Wang X, Bao P, et al. Platelet detection as a new liquid biopsy tool for human cancers. Front Oncol. 2022;12:983724.
Roweth HG, Battinelli EM. Lessons to learn from tumor-educated platelets. Blood. 2021;137(23):3174–80.
Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–9.
Climente-Gonzalez H, Porta-Pardo E, Godzik A, Eyras E. The functional impact of alternative splicing in Cancer. Cell Rep. 2017;20(9):2215–26.
Urbanski LM, Leclair N, Anczukow O. Alternative-splicing defects in cancer: splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip Rev RNA. 2018;9(4):e1476.
‘t Best MG. Veld S, Sol N, Wurdinger T. RNA sequencing and swarm intelligence-enhanced classification algorithm development for blood-based disease diagnostics using spliced blood platelet RNA. Nat Protoc. 2019;14(4):1206-34.
In ‘t Veld S, Arkani M, Post E, Antunes-Ferreira M, D’Ambrosi S, Vessies DCL, et al. Detection and localization of early- and late-stage cancers using platelet RNA. Cancer Cell. 2022;40(9):999–1009e6.
Oh SE, Seo JE, An JY, Lee JH, Sohn TS, Bae JM, et al. Prognostic impact of increased perioperative platelet count in gastric Cancer patients. J Surg Res. 2019;242:296–303.
Zhou Q, Huang F, He Z, Zuo MZ. Clinicopathological and prognostic significance of platelet count in patients with ovarian cancer. Climacteric. 2018;21(1):60–8.
Liu W, Ha M, Yin N. Combination of platelet count and lymphocyte to monocyte ratio is a prognostic factor in patients undergoing surgery for non-small cell lung cancer. Oncotarget. 2017;8(42):73198–207.
Nakahira M, Sugasawa M, Matsumura S, Kuba K, Ohba S, Hayashi T, et al. Prognostic role of the combination of platelet count and neutrophil-lymphocyte ratio in patients with hypopharyngeal squamous cell carcinoma. Eur Arch Otorhinolaryngol. 2016;273(11):3863–7.
Zhang F, Chen Z, Wang P, Hu X, Gao Y, He J. Combination of platelet count and mean platelet volume (COP-MPV) predicts postoperative prognosis in both resectable early and advanced stage esophageal squamous cell cancer patients. Tumour Biol. 2016;37(7):9323–31.
Gu L, Li H, Gao Y, Ma X, Chen L, Li X, et al. The association of platelet count with clinicopathological significance and prognosis in renal cell carcinoma: a systematic review and meta-analysis. PLoS ONE. 2015;10(5):e0125538.
Liu HB, Gu XL, Ma XQ, Lv TF, Wu Y, Xiao YY, et al. Preoperative platelet count in predicting lymph node metastasis and prognosis in patients with non-small cell lung cancer. Neoplasma. 2013;60(2):203–8.
Wang X, Ni X, Tang G. Prognostic role of platelet-to-lymphocyte ratio in patients with bladder Cancer: a Meta-analysis. Front Oncol. 2019;9:757.
Lusho S, Durando X, Mouret-Reynier MA, Kossai M, Lacrampe N, Molnar I, et al. Platelet-to-lymphocyte ratio is Associated with favorable response to Neoadjuvant Chemotherapy in Triple negative breast Cancer: a study on 120 patients. Front Oncol. 2021;11:678315.
Lee JW, Seol KH. Pretreatment Neutrophil-to-Lymphocyte Ratio Combined with Platelet-to-Lymphocyte Ratio as a Predictor of Survival Outcomes after Definitive Concurrent Chemoradiotherapy for Cervical Cancer. J Clin Med. 2021;10(10).
Takada K, Kashiwagi S, Asano Y, Goto W, Kouhashi R, Yabumoto A, et al. Prediction of Sentinel Lymph Node Metastasis using the platelet-to-lymphocyte ratio in T1 breast Cancer. Anticancer Res. 2020;40(4):2343–9.
Haghbin M, Hashemi Tayer A, Kamravan M, Sotoodeh Jahromi A. Platelet-derived Procoagulant Microparticles as blood-based biomarker of breast Cancer. Asian Pac J Cancer Prev. 2021;22(5):1573–9.
Tang M, Jiang L, Lin Y, Wu X, Wang K, He Q, et al. Platelet microparticle-mediated transfer of miR-939 to epithelial ovarian cancer cells promotes epithelial to mesenchymal transition. Oncotarget. 2017;8(57):97464–75.
Helley D, Banu E, Bouziane A, Banu A, Scotte F, Fischer AM, et al. Platelet microparticles: a potential predictive factor of survival in hormone-refractory prostate cancer patients treated with docetaxel-based chemotherapy. Eur Urol. 2009;56(3):479–84.
Da Silva JPA, Martins MR, Dos Santos RL, da Silva LM, Lima CAC, Torres LC, et al. Evaluation of platelet activation marker expression and its correlation with tumorigenesis and tumor progression in patients with gastric cancer. J Surg Oncol. 2022;126(1):125–31.
Ferroni P, Riondino S, Vazzana N, Santoro N, Guadagni F, Davi G. Biomarkers of platelet activation in acute coronary syndromes. Thromb Haemost. 2012;108(6):1109–23.
Farc O, Berindan-Neagoe I, Zaharie F, Budisan L, Zanoaga O, Cristea V. A role for serum cytokines and cell adhesion molecules in the non-invasive diagnosis of colorectal cancer. Oncol Lett. 2022;24(3):323.
Ilich A, Kumar V, Henderson M, Mallick R, Wells P, Carrier M, et al. Biomarkers in cancer patients at risk for venous thromboembolism: data from the AVERT study. Thromb Res. 2020;191(Suppl 1):31–S6.
Khorana AA, Ahrendt SA, Ryan CK, Francis CW, Hruban RH, Hu YC, et al. Tissue factor expression, angiogenesis, and thrombosis in pancreatic cancer. Clin Cancer Res. 2007;13(10):2870–5.
Uno K, Homma S, Satoh T, Nakanishi K, Abe D, Matsumoto K, et al. Tissue factor expression as a possible determinant of thromboembolism in ovarian cancer. Br J Cancer. 2007;96(2):290–5.
Normanno N, Cervantes A, Ciardiello F, De Luca A, Pinto C. The liquid biopsy in the management of colorectal cancer patients: current applications and future scenarios. Cancer Treat Rev. 2018;70:1–8.
Luo CL, Xu ZG, Chen H, Ji J, Wang YH, Hu W, et al. LncRNAs and EGFRvIII sequestered in TEPs enable blood-based NSCLC diagnosis. Cancer Manag Res. 2018;10:1449–59.
Xue L, Xie L, Song X, Song X. [Expression and significance of ACIN1 mRNA in platelets of Lung Cancer]. Zhongguo Fei Ai Za Zhi. 2018;21(9):677–81.
Yao B, Qu S, Hu R, Gao W, Jin S, Ju J, et al. Delivery of platelet TPM3 mRNA into breast cancer cells via microvesicles enhances metastasis. FEBS Open Bio. 2019;9(12):2159–69.
Asghar S, Waqar W, Umar M, Manzoor S. Tumor educated platelets, a promising source for early detection of hepatocellular carcinoma: liquid biopsy an alternative approach to tissue biopsy. Clin Res Hepatol Gastroenterol. 2020;44(6):836–44.
Waqar W, Asghar S, Manzoor S. Platelets’ RNA as biomarker trove for differentiation of early-stage hepatocellular carcinoma from underlying cirrhotic nodules. PLoS ONE. 2021;16(9):e0256739.
Wang H, Wei X, Wu B, Su J, Tan W, Yang K. Tumor-educated platelet miR-34c-3p and miR-18a-5p as potential liquid biopsy biomarkers for nasopharyngeal carcinoma diagnosis. Cancer Manag Res. 2019;11:3351–60.
Dong X, Song X, Ding S, Yu M, Shang X, Wang K et al. Tumor-educated platelet SNORD55 as a potential biomarker for the early diagnosis of non-small cell lung cancer. Thorac Cancer. 2021.
D’Ambrosi S, Visser A, Antunes-Ferreira M, Poutsma A, Giannoukakos S, Sol N, et al. The analysis of platelet-derived circRNA repertoire as potential diagnostic biomarker for Non-Small Cell Lung Cancer. Cancers (Basel). 2021;13:18.
Li X, Liu L, Song X, Wang K, Niu L, Xie L, et al. TEP linc-GTF2H2-1, RP3-466P17.2, and lnc-ST8SIA4-12 as novel biomarkers for lung cancer diagnosis and progression prediction. J Cancer Res Clin Oncol. 2021;147(6):1609–22.
Peterson JE, Zurakowski D, Italiano JE Jr, Michel LV, Connors S, Oenick M, et al. VEGF, PF4 and PDGF are elevated in platelets of colorectal cancer patients. Angiogenesis. 2012;15(2):265–73.
Mendoza-Almanza G, Burciaga-Hernandez L, Maldonado V, Melendez-Zajgla J, Olmos J. Role of platelets and breast cancer stem cells in metastasis. World J Stem Cells. 2020;12(11):1237–54.
Park CK, Kim JE, Kim MS, Kho BG, Park HY, Kim TO, et al. Feasibility of liquid biopsy using plasma and platelets for detection of anaplastic lymphoma kinase rearrangements in non-small cell lung cancer. J Cancer Res Clin Oncol. 2019;145(8):2071–82.
Sabrkhany S, Kuijpers MJE, Knol JC, Olde Damink SWM, Dingemans AC, Verheul HM, et al. Exploration of the platelet proteome in patients with early-stage cancer. J Proteom. 2018;177:65–74.
Lomnytska M, Pinto R, Becker S, Engstrom U, Gustafsson S, Bjorklund C, et al. Platelet protein biomarker panel for ovarian cancer diagnosis. Biomark Res. 2018;6:2.
Koduru SV, Tiwari AK, Hazard SW, Mahajan M, Ravnic DJ. Exploration of small RNA-seq data for small non-coding RNAs in human colorectal Cancer. J Genomics. 2017;5:16–31.
Zavesky L, Jandakova E, Weinberger V, Minar L, Hanzikova V, Duskova D, et al. Small non-coding RNA profiling in breast cancer: plasma U6 snRNA, miR-451a and miR-548b-5p as novel diagnostic and prognostic biomarkers. Mol Biol Rep. 2022;49(3):1955–71.
Acknowledgements
We owe thanks to the patients in our study and their family members. We really appreciate Dr. Zhaoyun Liu for her kind help in the figure preparation. The figures in the current study were created with BioRender.com.
Funding
This work was supported by the National Natural Science Foundation of China (81972014) and “Young Seedlings” Program of the Shandong Cancer Hospital Affiliated to Shandong First Medical University (CH-SFMU-QM20210005).
Author information
Authors and Affiliations
Contributions
SS Ding and XH Dong wrote the first draft, they contributed equally. XG Song designed and revised the manuscript; All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
Shanshan Ding, Xiaohan Dong, Xingguo Song declare no conflict of interest.
Ethical approval and Consent to participate
This study was approved by the Ethics Committee of Shandong Cancer Hospital Affiliated to Shandong First Medical University and Shandong Academy of Medical Sciences (2020001016).
Consent for publication
All the contributing authors and patients included in the study have consented to publish the paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ding, S., Dong, X. & Song, X. Tumor educated platelet: the novel BioSource for cancer detection. Cancer Cell Int 23, 91 (2023). https://doi.org/10.1186/s12935-023-02927-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-023-02927-5