
Abstract
Background
Prognostic assessment is imperative for clinical management of patients with hepatocellular carcinoma (HCC). Most reported prognostic signatures are based on risk scores summarized from quantitative expression level of candidate genes, which are vulnerable against experimental batch effects and impractical for clinical application. We aimed to develop a robust qualitative signature to assess individual survival risk for HCC patients.
Methods
Long non-coding RNA (lncRNA) pairs correlated with overall survival (OS) were identified and an optimal combination of lncRNA pairs based on the majority voting rule was selected as a classification signature to predict the overall survival risk in the cancer genome atlas (TCGA). Then, the signature was further validated in two external datasets. Besides, biomolecular characteristics, immune infiltration status, and chemotherapeutics efficacy of different risk groups were further compared. Finally, we performed key lncRNA screening and validated it in vitro.
Results
A signature consisting of 50 lncRNA pairs (50-LPS) was identified in TCGA and successfully validated in external datasets. Patients in the high-risk group, when at least 25 of the 50-LPS voted for high risk, had significantly worse OS than the low-risk group. Multivariate Cox, receiver operating characteristic (ROC) curve and decision curve analyses (DCA) demonstrated that the 50-LPS was an independent prognostic factor and more powerful than other available clinical factors in OS prediction. Comparison analyses indicated that different risk groups had distinct biomolecular characteristics, immune infiltration status, and chemotherapeutics efficacy. TDRKH-AS1 was confirmed as a key lncRNA and associated with cell growth of HCC.
Conclusions
The 50-LPS could not only predict the prognosis of HCC patients robustly and individually, but also provide theoretical basis for therapy. Besides, TDRKH-AS1 was identified as a key lncRNA in the proliferation of HCC. The 50-LPS might guide personalized therapy for HCC patients in clinical practice.
Similar content being viewed by others

Background
Liver cancer is one of the most lethal cancers worldwide and approximately 75–85% of the cases are diagnosed as hepatocellular carcinoma (HCC) pathologically [1]. For HCC patients with proper preserved liver function, curative surgical resection still remains the major treatment [2]. However, the prognosis of HCC patients varied tremendously after surgery due to tumor heterogeneity [2, 3], highlighting the need for personalized managements. Therefore, it is of critical importance to predict the prognosis of HCC patients after surgery for guiding clinical therapy and patient management.
Currently used clinical features and biomarkers, such as TNM staging system, BCLC staging system, and serum alpha-fetoprotein (AFP) level, were insufficient in providing accurate prognostic evaluation for HCC patients in clinical practice [4, 5]. For example, the most widely used serum AFP level was reported to be nearly half-negative in early and small size HCC patients [6], which crippled the prognostic ability of serum AFP. Moreover, the TNM and BCLC staging system were not widely used for the differences in etiology and genetic background of HCC patients [4].
Although many prognostic signatures based on scoring from quantitative gene expression have been developed for predicting prognosis of HCC patients recently [7,8,9], most of them require pre-collection of samples for data normalization and have inconsistent cut-off value in different cohorts, which are impractical for clinical application. Conversely, the within-sample relative expression orderings (REOs) of gene pairs, which is an individual qualitative transcriptional trait, has been demonstrated to be robust against batch effects and data normalization [10, 11]. Based on the REOs, some qualitative signatures were created and achieved satisfactory performance in prognosis prediction for several cancers [12,13,14,15]. However, most reported qualitative prognostic signatures were based on protein-coding genes and few of them focused on HCC. Considering the important role of long noncoding RNA (lncRNA) in the carcinogenesis of HCC [16], it is worthy to find an individualized lncRNA prognostic signature for HCC patients after surgery.
In this study, a novel REOs-based prognostic signature consisting of 50 lncRNA pairs (50-LPS) was identified in a training dataset, and the signature was successfully validated in two external datasets. We further revealed the different biomolecular characteristics, immune infiltration status, and chemotherapeutics efficacy between the two prognostic groups. Moreover, among the 55 lncRNAs included in the 50-LPS, TDRKH-AS1 was confirmed as a key lncRNA and associated with cell growth of HCC. In clinical practice, these results might be useful for aiding personalized therapy and management of HCC patients.
Methods
Data source and preprocessing
All the cohorts of HCC samples, as described in Additional file 1: Table S1, were downloaded from public resources. Gene expression profile and somatic mutation data of the TCGA-LIHC cohort were obtained by using R package TCGAbiolinks [17] and TCGAmutations [18] respectively. Clinical information of the TCGA-LIHC cohort was downloaded from UCSC Xeno (https://xena.ucsc.edu/public) on 2019-11-10. Transcriptome data of CHCC cohort was accessed from NODE (https://www.biosino.org/node) and the corresponding clinical information was acquired from a previous study [19]. Transcriptome and clinical information of LIRI samples, included in the PCAWG project, were downloaded from ICGC (https://dcc.icgc.org/releases/PCAWG) [20]. GSE77509 and GSE104580 were downloaded from the Gene Expression Omnibus (GEO) database. After mapping the ensemble gene IDs to gene names according to the annotation files (hg38.99) downloaded from Ensemble (http://asia.ensembl.org), all the gene expression values were transformed into transcripts per million (TPM) values. LncRNAs were kept for further analysis, whose expression value was more than zero in more than 90% of samples both in TCGA-LIHC and CHCC cohorts.
Survival analysis
The overall survival (OS) interval was defined as the time from surgery to death or the last follow-up. Kaplan–Meier survival curves and log-rank test were used to evaluate the difference of OS between the high-risk and low-risk subgroups. The univariate Cox regression model was utilized to identify prognostic lncRNA pairs and clinical factors for HCC patients. The multivariate Cox regression model was used to evaluate prognostic performance of the signature after adjusting for age, gender, serum AFP levels, liver cirrhosis, tumor vascular invasion (VI), and TNM stage. Hazard ratios (HRs) and 95% confidence intervals (CIs) were calculated from the Cox proportional- hazards model. The predictive ability of the signature was evaluated by the concordance index (C-index) value and the AUC value of the ROC curve [21]. Decision curve analysis (DCA) was used to assess the utility of the signature for decision making [22].
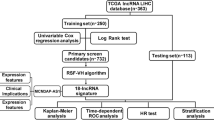
Identification of the signature
First, lncRNAs with significant prognostic value were identified by using univariate Cox regression analyses in the training dataset (TCGA-LIHC). Then, lncRNA pairs were constructed via permutation and combination by R software. For a given lncRNA pair, for example, lncRNA 1 and lncRNA 2 with expression levels of E1 and E2, its REO pattern (E1 > E2 or E1 < E2) can stratify all samples into two groups. If the two groups have significantly different OS, the lncRNA pair is considered as a candidate prognostic lncRNA pair. Next, a forward selection procedure was adopted to seek an optimal subset of the lncRNA pairs, which was based on the pre-defined classification rule: a patient is classified into the high-risk group if no less than half of the lncRNA pairs of this patient vote for high risk; otherwise, the low-risk group. In brief, the candidate lncRNA pair with the highest C-index was selected as the seed signature, and the other prognosis‐related lncRNA pairs, ranked in descending C-index order, were added into the signature one by one. Every time an additional lncRNA pair added in the signature, a new C-index value was calculated to check if the predictive ability of the signature was better than before. Finally, the optimal REO lncRNA signature was identified when an addition of a new lncRNA pair did not improve the C-index of the signature in the training dataset.
Analyses of transcriptional and genomic data
The Wilcoxon rank-sum test was performed to identify differentially expressed genes between the low- and high-risk subgroups. Gene Set Enrichment Analysis (GSEA) was conducted by using clusterProfiler R package [23] to explore the difference of biological characteristics between the two groups. Genes with different mutation frequencies between the two prognostic groups were visualized by complexheatmap R package [24].
Estimation of immune infiltration
CIBERSORT algorithm [25] was adopted to investigate the immune-cell infiltration. The differences of immune infiltrating cell scores were analyzed by Wilcoxon rank-sum test.
Characterization of HCC subclasses
To analyze the correlation between the two prognostic subgroups and previously published HCC molecular subtypes, MS.liverK algorithm [26] was used to characterize the six different molecular subtypes in TCGA-LIHC samples. Chi-square test was used to detect correlations between the two subgroups and HCC subclasses.
Prediction of the benefit of each risk group from chemotherapy
Our previous data (GSE104580) consisting of HCC patients treated with transarterial chemoembolization (TACE) was used to indirectly predict the chemotherapy efficacy of the two risk subgroups based on subclass mapping analysis [27]. In addition, to test whether the subgroup could benefit from other chemotherapeutic drugs, we downloaded the predicted half maximal inhibitory concentration (IC50) data of TCGA-LIHC samples from a previous study [28]. Wilcoxon rank-sum test was used to detect the difference of predicted IC50 value between the two groups.
Hub lncRNAs screening and validation in vitro
Hub lncRNAs were defined as the differentially expressed lncRNAs with log2(fold change) > 1 and FDR < 0.05 (Wilcoxon test) between tumor and normal liver tissues, as well as associated with OS in TCGA-LIHC cohort and CHCC cohort. To validate the expression pattern between tumor and normal liver tissues by quantitative real‑time PCR, we collected five paired HCC and normal liver tissues in our center. This research was approved by the Ethics Committee of Sun Yat-Sen University Cancer Center, and written informed consent was obtained from all patients. In addition, we conducted in vitro assays to investigate the biological function of the key lncRNA.
RNA extraction, reverse transcription, and quantitative real‑time PCR
According to the manufacturer’s instructions, total RNA was isolated by using an RNA Extraction Kit (ESscience Biotech, Shanghai, China). For reverse transcription, 2 μg of total RNA was used to synthesize cDNA with a cDNA Synthesis Kit (TOYOBO, Osaka, Japan). Then cDNA was subjected to quantitative real‑time PCR amplification using SYBR Green (TOYOBO, Osaka, Japan) with a Bio-Rad PCR System. β-actin was used as an internal control. The sequence of primers used in this study was provided in Additional file 1: Table S2.
Cell lines and culture conditions
HCC cell lines, MHCC97H and Huh7, were obtained from Shanghai Municipal Liver Cancer Medical Center and the National Collection of Authenticated Cell Cultures, respectively. The cell lines were incubated in Dulbecco’s modified Eagle’s medium (Gibco, Carlsbad, USA) with 10% foetal bovine serum (Gibco, Carlsbad, USA), with 5% CO2 at 37 °C.
Plasmid construction and transfection
The pSLenti vector containing short hairpin RNAs (shRNA) targeting TDRKH-AS1 was obtained from OBiO Technology (OBiO, Shanghai, China). According to the instructions, plasmid was transfected into MHCC97H and Huh7 cell line using a Lenti-Pac™ HIV Expression Packaging Kit (GeneCopoeia, Rockville, USA). The sequence of the shRNA was provided in Additional file 1: Table S2.
In vitro cell growth assays and apoptosis assays
For CCK8 assays, the transfected cells were plated in 96-well plates at a density of 2000 cells per well. Optical Density (OD) value at 450 nm was tested on day 0, 1, 2, 3, and 4 with a CCK8 assay kit (Dojindo, Kumamoto, Japan). For Cell colony formation assays, the transfected cells were plated in 6-well plates at a density of 1000 cells per well and harvested on day 14. Crystal violet staining solution (Beyotime, Shanghai, China) was used to stain the cell colony. The total area of the colony was detected by Image J software (NIH Image, Bethesda, MD). According to the manufacturers’ instructions, apoptosis assays were performed by PI/Annexin V detection kits (ESscience Biotech, Shanghai, China) and analyzed by flow cytometry (Beckman CytoFLEX, USA).
Western blotting
Cell extracts were harvested and were resolved by SDS-PAGE and transferred to a polyvinylidene difluoride membrane. After blocking, the membrane was incubated with primary antibodies including Caspase3 (1:1000, Cell Signaling Technology, USA), phospho-Akt (Ser473)(1:1000, Cell Signaling Technology, USA), GAPDH (1:1000, Cell Signaling Technology, USA) and the secondary antibody HRP-conjugated goat anti-rabbit IgG (1:3000; Cell Signaling Technology, USA). Signals were detected by an ECL kit (Bio-Rad, USA).
Statistical analysis
All the computational and statistical analyses were performed using R 4.0.1 or GraphPad Prism 8.0.1 software. P-value < 0.05 was regarded as statistically significant. The false discovery rate (FDR) was calculated using the Benjamini–Hochberg method [29] when multiple testing occurred.
Results
Development of the REOs-based prognostic signature
The main research steps of this study are summarized in Fig. 1. TCGA-LIHC cohort with 365 HCC samples was used as the training dataset. By setting a cut-off less than 0.05 of FDR in the univariate Cox proportional hazards regression model, we identified 119 OS-associated lncRNAs. For all the lncRNA pairs consisting of every two of the 119 pre-selected lncRNAs, 1333 prognosis‐associated lncRNA pairs were chosen (FDR < 0.2) and then ranked in descending order according to their C-index values. According to the majority voting rule (see “Methods” section), a final set of 50 lncRNA pairs (Table 1) attained the highest C-index value of 0.75 was identified through the forward selection procedure (see “Methods” section).

Overview of the study workflow
Using the 50 lncRNA pairs signature, named as 50-LPS, in the training cohort, 135 samples were stratified into high-risk group, who got no less than 25 lncRNA pairs voting for high risk, and other samples were stratified into low-risk group. KM plot showed that samples in the high-risk group had an obviously worse overall survival than those in the low-risk group (HR = 5.92, 95% CI 4.09–8.56, p = 3.56 × 10–26, Fig. 2A). ROC curve indicated that the 50-LPS had a robust predictive ability with an AUC value of 0.83, 0.77, 0.74 in the 1st year, 3rd year and 5th year (see Fig. 2D), respectively. What’s more, the 50-LPS maintained a significant prognostic power after adjusting for age, gender, liver cirrhosis, serum AFP level, tumor vascular invasion (VI) and TNM stage (Fig. 2E) and presented a better utility than TNM stage and VI in DCA (Fig. 2F). Peculiarly, the 50-LPS can successfully stratify the early-stage (stage I + II) samples into high- and low-risk group with significant OS difference (HR = 6.45, 95% CI 3.90–10.66, p = 1.47 × 10–16, Fig. 2B) as well as the late-stage (stage III + IV) samples (HR = 5.1, 95% CI 2.54–10.24, p = 5.21 × 10–7, Fig. 2C).

The overall survival outcome of the two risk subgroups stratified by the 50-LPS in the training cohort (TCGA-LIHC). The Kaplan–Meier curves of overall survival for the entire training cohort (A) and patients in TNM I + II (B) and TNM III + IV (C). The time-dependent ROC analysis of the 50-LPS in the training cohort (D). Univariate and multivariate Cox regression analyses of OS in the training cohort (E). DCA of prognostic factors identified from univariate cox in training cohort (F)
Validation of the signature
Two external HCC cohorts were used to test the predictive ability and utility of the 50-LPS. A number of 159 HBV associated HCC samples in CHCC cohort were firstly subjected to risk assessment by using the 50-LPS. 69 and 90 samples were classified into the high- and low-risk groups, respectively. The high-risk group had a significantly shorter OS when compared to the low-risk group (HR = 3.22, 95% CI 1.85–5.61, p = 1.32 × 10–5, Fig. 3A) with an AUC value of 0.66 and 0.64 at 1st year and 3rd year respectively (Fig. 3G). The 42 samples from LIRI cohort were used as the second validation set. 13 and 29 samples were predicted to be in the high- and low-risk groups with extremely different OS (HR = 6.87, 95% CI 2.03–23.2, p = 3.57 × 10–4, Fig. 3D) with an AUC value of 0.87 and 0.77 at 1st year and 3rd year respectively (Fig. 3J). In addition, the 50-LPS also succeeded in stratifying the early-stage or late-stage HCC patients from CHCC cohort (Fig. 3B, C) and LIRI cohort (Fig. 3E, F).

Performance of the 50-LPS in two external cohorts. The Kaplan–Meier curves of overall survival for the entire CHCC cohort (A) and TNM I + II (B) and TNM III + IV (C) patients. The Kaplan–Meier curves of overall survival for the entire LIRI cohort (D) and TNM I + II (E) and TNM III + IV (F) patients. The time-dependent ROC analysis of the 50-LPS in the CHCC cohort (G) and LIRI cohort (J). Univariate and multivariate Cox regression analyses of OS in the CHCC cohort (H) and LIRI cohort (K). DCA of prognostic factors identified from univariate cox in CHCC cohort (I) and LIRI cohort (L)
Furthermore, the multivariate Cox regression analysis, ROC curve and DCA were also performed in the two validation cohorts. As expected, we found that the 50-LPS exhibited a consistent powerful ability and utility for OS-prediction and dwarfed other clinical factors including TNM stage, serum AFP level, and VI (Fig. 3H, I, K, L).
Distinct transcriptional and genomic characteristics of the two prognostic groups
In order to better characterize the biomolecular characteristics of the two risk-groups, differential analyses of their transcriptome and somatic mutation data were conducted. We obtained the differential expressed genes between the two prognostic groups and performed GSEA to identify pathways enriched in each subgroup. As shown in Fig. 4A, the high-risk group typically enriched in some pathways relevant to tumor proliferation and metastasis such as G2M checkpoint, E2F targets, epithelial-mesenchymal transition (EMT), while the low-risk group mainly enriched in several metabolism-relevant pathways including fatty acid metabolism and bile acid metabolism.

Biological features and immune cell infiltration of the two risk subgroups stratified by the 50-LPS. Pathway enrichment analysis by GSEA (A). The difference of immune cell infiltrated in the tumor microenvironment between the two groups (B). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
To further investigate the differences in somatic mutation frequency between the two subgroups, we analyzed genes with high mutation frequency or in important pathways relevant to HCC, including P53, cell cycle pathway, WNT beta-catenin pathway, and hepatic differentiation. Results in Fig. 5B showed that the high-risk group displayed a higher mutation frequency of TP53 (46%) than the low-risk group (21%), implying the dysregulation of cell proliferation in high-risk group.

Clinical, molecular and genomic characteristics of the two risk subgroups stratified by the 50-LPS. Clinical characteristics of HCC subgroups and association with previous HCC molecular subtypes (A). The difference of frequencies in gene mutation between the two groups (B). *p < 0.05, **p < 0.01, ***p < 0.001
Difference of immune infiltration landscape in two HCC subgroups
Because recent study indicated that immunologic background may influence the prognostic result of HCC patients, the difference of the immune cell infiltration between the two prognostic groups deserves further elaboration. Using the CIBERSORT algorithm, we revealed that the low-risk group had a relatively higher level of tumor-infiltrating immune cells including naïve B cells, CD8+ T cells, monocytes, M1 macrophages and resting Mast cells, while the high-risk group had a richer infiltration of follicular helper cells, regulatory T cells, M0 macrophages and neutrophils (Fig. 4B).
Correlation of the HCC subgroups with clinical characteristics and previously reported subclasses
We explored the relationship between the clinicopathological features and HCC subgroups classified by our novel signature in the TCGA-LIHC cohort. As illustrated in the heatmap (Fig. 5A), samples in the two subgroups had several different clinicopathological characteristics. According to the result of chi-square test, the high-risk group was correlated with advanced pathologic stage (p < 0.001), high serum AFP level (p = 0.003), and more frequent VI situation (p = 0.002), but displayed no difference with the low-risk group in age, gender and liver cirrhosis.
What’s more, we compared our classification with previously published HCC molecular subclasses to better understand the molecular background underlying the two prognostic subgroups. The heatmap (Fig. 5A) revealed that the high-risk group was obviously linked to Hoshida’s [30] S1/S2 subclass (p < 0.001), Lee’s [31] A subclass (p < 0.001), Roessler’s [32] subgroup A(p < 0.001), Chiang’s [33] proliferation (p < 0.001) class and Boyault’s [34] G2/G3 (p < 0.001), whereas the low-risk group was significantly correlated with Hoshida’s S3 (p < 0.001), Lee’s B subtype (p < 0.001), Chiang’s CTNNB1 (p < 0.001) class and Boyault’s G4/G5 subclass(p < 0.001).
Correlation between the signature and chemotherapeutics
Since the two groups of patients with different OS outcomes have different biological characteristics, we then wanted to find out whether there was a difference in chemotherapeutic drug sensitivity between them.
Using the subclass mapping method, we compared the transcriptome profile of the two subgroups with our previous cohort containing 147 HCC patients who received TACE. Surprisingly, we found that the low-risk group was significantly correlated with the TACE-response group (p < 0.001, Fig. 6A), while the high-risk group showed an obvious similarity to the TACE-resistant group (p < 0.001, Fig. 6A), which indicated that the low-risk group but not the high-risk group would benefit from TACE therapy.

Correlations of the HCC subgroups with the response to different chemotherapeutics. Subclass mapping correlation analysis between HCC subgroups and samples with different sensitivities to TACE (A), and several anticancer drugs (B). TACE-NR, TACE non-responder; TACE-R, TACE responder. ** p<0.01,*** p<0.001,**** p<0.0001
In addition, we also explored the efficacy of different antitumor agents between the two groups. The result of Wilcoxon rank-sum test revealed that the low-risk group had a lower predicted half maximal inhibitory concentration (IC50) of sorafenib, vincristine and methotrexate, whereas the high-risk group had lower predicted IC50 of gemcitabine, epirubicin and etoposide (Fig. 6B), which meant that this signature could be a promising indicator for chemosensitivity prediction.
Hub lncRNA screening identified TDRKH-AS1 as a key lncRNA in HCC
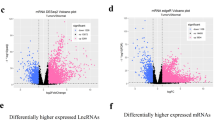
To screen out the hub lncRNAs included in the 50-LPS, we performed differential expression analysis as well as survival analysis in multiple HCC cohorts. In the 50 tumor samples with paired normal liver tissue from TCGA-LIHC cohort, we found that most of the lncRNAs included in the 50-LPS were upregulated in tumor compared with normal liver (Fig. 7A). Especially, ten lncRNAs were significantly upregulated (log2FC > 1, FDR < 0.05, Fig. 7B), implying the potential function role of these lncRNAs in the carcinogenesis of HCC. Next, we further tested the expression pattern of these ten lncRNAs in CHCC cohort and GSE77509, which included 159 HCC samples and 20 HCC samples with paired normal liver tissue, respectively. Moreover, the prognostic significance of them was also evaluated in TCGA-LIHC and CHCC cohort.

Hub lncRNA screening identified TDRKH-AS1 as a key lncRNA in HCC. Expression heatmap of the 55 lncRNAs included in the 50-LPS in the 50 paired tumor and normal liver tissues of TCGA-LIHC cohort (A). Volcano plot of the differential test and the top 6 differentially expressed lncRNAs with log2FC > 1 and FDR < 0.05 were marked in orange (B). Venn diagram identified two lncRNAs (TDRKH-AS1 and MAFG-DT) that were differentially expressed in TCGA-LIHC, CHCC and GSE77509, as well as associated with overall survival in TCGA-LIHC and CHCC cohort (C). The Kaplan–Meier curves of overall survival for TDRKH-AS1 and MAFG-DT in TCGA-LIHC (D, E). Real-time PCR validated the expression difference of TDRKH-AS1 and MAFG-DT in five paired HCC and normal liver tissues (F). *p < 0.05, **p < 0.01, ***p < 0.001. T, tumor; N, normal
As shown in Fig. 7C–E, two lncRNAs, TDRKH-AS1 and MAFG-DT, were consistently upregulated in three cohorts and associated with poor survival in TCGA-LIHC and CHCC cohort. We further validated the expression pattern of the two lncRNAs in 5 paired HCC and normal liver tissues from our center by using quantitative real-time PCR. As expected, TDRKH-AS1 and MAFG-DT were both highly expressed in HCC than normal liver (p < 0.01, Fig. 7F). However, TDRKH-AS1 was more abundant in relative expression than MAFG-DT, indicating a more essential role of TDRKH-AS1 in HCC tumor biology. Therefore, TDRKH-AS1 was considered as the hub lncRNA for further function investigation.
Validation of the biological function of TDRKH-AS1 in HCC
To validate the biological function of TDRKH-AS1 in HCC, we performed loss-of-function assays in HCC cell lines, MHCC97H and Huh7. After knocking down the expression of TDRKH-AS1 using short hairpin RNAs (shRNA) (p < 0.01 in Huh7, p < 0.001 in MHCC97H, Fig. 8A), we conducted CCK8 and colony formation assays to detect the influence of TDRKH-AS1 on cell growth of HCC in vitro. As shown in the CCK8 assay (p < 0.001 in Huh7, p < 0.01 in MHCC97H, Fig. 8B, C), the cell growth rate of both cell lines was significantly decreased after knocking down TDRKH-AS1. Restoration of TDRKH-AS1 expression in Huh7 cell line could restore cell growth (p<0.001, Additional file 1: Fig. S1 A, B). Similarly, the result of colony formation assay revealed that the proliferation ability of both cell lines was significantly hampered when TDRKH-AS1 was knocked down (p < 0.01 in Huh7, p < 0.01 in MHCC97H, Fig. 8D, E). In addition, the percentage of apoptotic cells were remarkably increased (p < 0.001 in Huh7, p < 0.001 in MHCC97H, Fig. 8F, G, H), and the pro-apoptotic protein, cleaved caspase3, was increased after knockdown of TDRKH-AS1 (Fig. 8I). Moreover, we found that the level of Akt phosphorylation on Ser 473 was decreased (Fig. 8I). These results suggested that TDRKH-AS1 might be essential for tumor growth and cell survival of HCC.

Validation of the biological function of TDRKH-AS1 in HCC. TDRKH-AS1 was successfully knocked down in Huh7 and MHCC97H cells (A). CCK8 assay in Huh7 (B) and MHCC97H (C) after knockdown of TDRKH-AS1. Colony formation assay in Huh7 and MHCC97H after knockdown of TDRKH-AS1 (D, E). Apoptosis assay in Huh7 and MHCC97H after knockdown of TDRKH-AS1 (F–H). Cleaved caspase3 and phospho-Akt (Ser473) were detected in Huh7 and MHCC97H by western blotting after knockdown of TDRKH-AS1 (I). *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
In this study, we developed a novel individualized lncRNA signature, 50-LPS, to predict the risk of prognosis in patients with hepatocellular carcinoma after surgery. This signature was successfully validated in two external datasets and displayed high stability and robust predictive ability in all three datasets. Moreover, it was more reliable than serum AFP level, tumor vascular invasion, cirrhosis and TNM stage for OS prediction. Our study provided a novel prognostic indicator for clinical application.
As a qualitative signature, the 50-LPS exhibited a better feasibility than quantitative risk score that mostly reported. Since it was designed by using the majority voting rule based on the REOs trait of lncRNA pairs within a single sample, it did not require pre-collection of massive samples for data normalization and cut-off determination. When using the 50-LPS, the relative expression rank of lncRNAs within a single sample can be obtained by RNA sequencing, microarray or mere quantitative polymerase chain reaction, without considering sequencing depth or batch influence. Therefore, the 50-LPS can be easily applied personally. Ao et al. [35] reported a qualitative prognostic signature consisting of 20 gene-pairs for HCC and proved that qualitative signatures were reliable in prognosis prediction. However, Ao’s signature only focused on the patients with early-stage HCC and protein-coding genes. Our study revealed that qualitative signatures derived from lncRNA pairs were also powerful and feasible in prognosis prediction for HCC in early stage as well as advanced stage. A target panel will be designed for clinical translational research in our next work.
Intriguingly, the high- and low-risk groups identified by the 50-LPS was, to some extent, in agreement with previously reported molecular subtypes [9] of HCC despite of different methods and criteria used. It supports the notion that HCC patients from the two risk groups stratified by the 50-LPS might represent distinct disease entities. We observed that HCC patients in the high-risk group were biologically deficient in p53 pathway and active in E2F pathway and EMT pathway. Clinically, patients in the high-risk group exhibited more aggressive traits with high AFP level, poor tumor differentiation, vascular invasion, advanced cancer stages, and poor prognosis. As a contrast, the low-risk subclass showed retained hepatocyte-like phenotype, with moderate-high tumor differentiation, and better prognosis. In addition, nearly half of the high-risk patients had TP53 mutation. TP53 mutation could cause genomic instability and uncontrolled cell proliferation [36, 37], which might partially account for the poor OS outcome of the high-risk group. However, there is still a variation in the proportion of each risk group from different cohorts, which might result from the regional etiology difference or genetic background difference.
Although immunotherapy offers new promise to patients with HCC [38], there is still a lack of validated biological markers for predicting the therapy efficacy and guiding clinical decision-making [39]. In our study, tumor tissues in the low-risk group were higher in CD8+ T cell infiltration than the high-risk group, indicating that the tumor immune microenvironment in the low-risk tumor was in a more activated status. Patients from the low-risk group might be more likely to benefit from the therapy of immune checkpoint inhibitors (ICIs), yet it needs future validation. In addition, the submap results prompted that HCC patients in the low-risk group might be more likely to respond to TACE therapy, which is the standard treatment for intermediate-stage HCC [40]. Apart from that, we found that the 50-LPS could predict the efficacy of several common anticancer drugs, including sorafenib, vincristine, methotrexate, epirubicin, etoposide and gemcitabine. Taken together, the 50-LPS may be used as a tool together with other indicators in evaluating the efficacy of ICIs, TACE and some anticancer drugs, which may help improve the dilemmas in the precision treatment of HCC.
Our study has identified tens of OS-related lncRNAs in HCC and demonstrated in vitro that TDRKH-AS1 could influence the cell growth of HCC. A previous study reported that lncRNA TDRKH-AS1 could target β-catenin in the Wnt signaling pathway to promote colorectal cancer cell proliferation and invasion [41]. However, our study revealed that knockdown of TDRKH-AS1 might influence cell proliferation by inducing apoptosis in HCC. Our future researches will focus on the molecular signal pathway that TDRKH-AS1 may trigger in HCC, which would help us understand the intrinsic mechanism determining different prognosis of HCC patients more comprehensively.
Certainly, there are several limitations in our study that need to be improved. A major limitation is that the training dataset and validation datasets are all from public databases. A prospective study will be needed to validate the effectiveness and utility of the 50-LPS. Additionally, in order to improve the accuracy of OS prediction, the 50-LPS could be used in combination with other clinicopathologic features, such as AFP, VI and pathological stage in the future.
Conclusions
In summary, current study developed a novel individualized lncRNA signature, 50-LPS, that could not only predict the prognosis of HCC patients robustly but also provide theoretical basis for precision therapy. Additionally, TDRKH-AS1 was identified as a key lncRNA in the proliferation of HCC. The 50-LPS might help guide personalized therapy for HCC patients in clinical practice.
Availability of data and materials
TCGA-LIHC can be downloaded from UCSC Xeno (https://xena.ucsc.edu/public). CHCC cohort can be accessed from NODE (https://www.biosino.org/node, ID# OEP000321). LIRI samples included in the PCAWG project can be downloaded from ICGC (https://dcc.icgc.org/releases/PCAWG). GSE77509 and GSE104580 are accessible in Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo).
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- OS:
-
Overall survival
- lncRNA:
-
Long non-coding RNA
- 50-LPS:
-
50 LncRNA pairs
- REOs:
-
Relative expression orderings
- TCGA:
-
The cancer genome atlas
- GEO:
-
Gene expression omnibus
- PCAWG:
-
Pan-cancer analysis of whole genomes
- TNM:
-
Tumor node metastasis
- BCLC:
-
Barcelona clinic liver cancer
- CHCC:
-
Chinese hepatocellular carcinoma
- LIRI:
-
Liver cancer RIKEN
- ICGC:
-
International cancer genome consortium
- LIHC:
-
Liver hepatocellular carcinoma
- FPKM:
-
Fragment per kilobase million
- TPM:
-
Transcripts per million
- DCA:
-
Decision curve analysis
- ROC:
-
Receiver operating characteristic curve
- AUC:
-
Area under curve
- CI:
-
Confidence interval
- FDR:
-
False discovery rate
- EMT:
-
Epithelial mesenchymal transition
- GSEA:
-
Gene get enrichment analysis
- VI:
-
Vascular invasion
- TACE:
-
Transarterial chemoembolization
- IC50:
-
Half maximal inhibitory concentration
- ICIs:
-
Immune checkpoint inhibitors
- AFP:
-
Alpha-fetoprotein
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–14.
Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380(15):1450–62.
Subramaniam S, Kelley RK, Venook AP. A review of hepatocellular carcinoma (HCC) staging systems. Chin Clin Oncol. 2013;2(4):33.
Toyoda H, Kumada T, Tada T, Sone Y, Kaneoka Y, Maeda A. Tumor markers for hepatocellular carcinoma: simple and significant predictors of outcome in patients with HCC. Liver Cancer. 2015;4(2):126–36.
Wang T, Zhang KH. New blood biomarkers for the diagnosis of AFP-negative hepatocellular carcinoma. Front Oncol. 2020;10:1316.
Yang C, Huang X, Li Y, Chen J, Lv Y, Dai S. Prognosis and personalized treatment prediction in TP53-mutant hepatocellular carcinoma: an in silico strategy towards precision oncology. Brief Bioinform. 2021. https://doi.org/10.1093/bib/bbaa164.
Zhang B, Tang B, Gao J, Li J, Kong L, Qin L. A hypoxia-related signature for clinically predicting diagnosis, prognosis and immune microenvironment of hepatocellular carcinoma patients. J Transl Med. 2020;18(1):342.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J, Finn RS. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6.
Zheng H, Song K, Fu Y, You T, Yang J, Guo W, Wang K, Jin L, Gu Y, Qi L, et al. An absolute human stemness index associated with oncogenic dedifferentiation. Brief Bioinform. 2021;22(2):2151–60.
Ao L, Zhang Z, Guan Q, Guo Y, Guo Y, Zhang J, Lv X, Huang H, Zhang H, Wang X, et al. A qualitative signature for early diagnosis of hepatocellular carcinoma based on relative expression orderings. Liver Int. 2018;38(10):1812–9.
Qi L, Li T, Shi G, Wang J, Li X, Zhang S, Chen L, Qin Y, Gu Y, Zhao W, et al. An individualized gene expression signature for prediction of lung adenocarcinoma metastases. Mol Oncol. 2017;11(11):1630–45.
Zheng H, Song K, Fu Y, You T, Yang J, Guo W, Wang K, Jin L, Gu Y, Qi L, et al. A qualitative transcriptional signature for determining the grade of colorectal adenocarcinoma. Cancer Gene Ther. 2020;27(9):680–90.
Huang H, Zou Y, Zhang H, Li X, Li Y, Deng X, Sun H, Guo Z, Ao L. A qualitative transcriptional prognostic signature for patients with stage I-II pancreatic ductal adenocarcinoma. Transl Res. 2020;219:30–44.
Yang J, Song K, Guo W, Zheng H, Fu Y, You T, Wang K, Qi L, Zhao W, Guo Z. A qualitative transcriptional signature for predicting prognosis and response to bevacizumab in metastatic colorectal cancer. Mol Cancer Ther. 2020;19(7):1497–505.
Huang Z, Zhou JK, Peng Y, He W, Huang C. The role of long noncoding RNAs in hepatocellular carcinoma. Mol Cancer. 2020;19(1):77.
Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. TCGAbiolinks: an R/bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44(8):e71–e71.
Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018;6(3):271-281 e277.
Gao Q, Zhu H, Dong L, Shi W, Chen R, Song Z, Huang C, Li J, Dong X, Zhou Y, et al. Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell. 2019;179(2):561-577 e522.
Goldman MJ, Zhang J, Fonseca NA, Cortes-Ciriano I, Xiang Q, Craft B, Pineiro-Yanez E, O’Connor BD, Bazant W, Barrera E, et al. A user guide for the online exploration and visualization of PCAWG data. Nat Commun. 2020;11(1):3400.
Van Oirbeek R, Lesaffre E. An application of Harrell’s C-index to PH frailty models. Stat Med. 2010;29(30):3160–71.
Vickers AJ, Cronin AM, Elkin EB, Gonen M. Extensions to decision curve analysis, a novel method for evaluating diagnostic tests, prediction models and molecular markers. BMC Med Inform Decis Mak. 2008;8:53.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–9.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7.
Petitprez F, Meunier L, Letouzé E, Hoshida Y, Villanueva A, Llovet J, Thorgeirsson S, Wang XW, Fridman WH, Zucman-Rossi J, et al. MS.liverK: an R package for transcriptome-based computation of molecular subtypes and functional signatures in liver cancer. BioRxiv. 2019. https://doi.org/10.1101/540005.
Hoshida Y, Brunet JP, Tamayo P, Golub TR, Mesirov JP. Subclass mapping: identifying common subtypes in independent disease data sets. PLoS ONE. 2007;2(11):e1195.
Li Y, Umbach DM, Krahn JM, Shats I, Li X, Li L. Predicting tumor response to drugs based on gene-expression biomarkers of sensitivity learned from cancer cell lines. BMC Genomics. 2021;22(1):272.
Benjamini YHY. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc. 1995;57:289–300.
Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69(18):7385–92.
Lee JS, Chu IS, Heo J, Calvisi DF, Sun Z, Roskams T, Durnez A, Demetris AJ, Thorgeirsson SS. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40(3):667–76.
Roessler S, Jia HL, Budhu A, Forgues M, Ye QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX, et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010;70(24):10202–12.
Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, LeBlanc AC, Donovan DJ, Thung SN, Sole M, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68(16):6779–88.
Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, Herault A, Saric J, Belghiti J, Franco D, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45(1):42–52.
Ao L, Song X, Li X, Tong M, Guo Y, Li J, et al. An individualized prognostic signature and multi-omics distinction for early stage hepatocellular carcinoma patients with surgical resection. Oncotarget. 2016;7(17):24097–110.
Levine AJ. Spontaneous and inherited TP53 genetic alterations. Oncogene. 2021;40(41):5975–83.
Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer. 2020;20(8):471–80.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–905.
Llovet JM, Castet F, Heikenwalder M, Maini MK, Mazzaferro V, Pinato DJ, Pikarsky E, Zhu AX, Finn RS. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2021. https://doi.org/10.1038/s41571-021-00573-2.
Llovet JM, De Baere T, Kulik L, Haber PK, Greten TF, Meyer T, Lencioni R. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2021;18(5):293–313.
Jiao Y, Zhou J, Jin Y, Yang Y, Song M, Zhang L, Zhou J, Zhang J. Long non-coding RNA TDRKH-AS1 promotes colorectal cancer cell proliferation and invasion through the beta-catenin activated Wnt signaling pathway. Front Oncol. 2020;10:639.
Acknowledgements
We would like to thank TCGA, GEO, PCAWG and NODE databases for their contribution.
Funding
This research was funded by National Natural Science Foundation of China (81625017) and National Natural Science Foundation of China (82072610).
Author information
Authors and Affiliations
Contributions
Conception and design: MS and XYB; collection and assembly of data: XYB, LYM and SL; data analysis and interpretation: XYB and SL; manuscript writing: XYB, XJL and YJX; manuscript editing: AK, DSW and SL. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research was approved by the Ethics Committee of Sun Yat-Sen University Cancer Center.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Table S1. Description of the datasets analyzed in this study. Table S2. Sequences of primers and short hairpin RNAs. Fig. S1. Restoration of TDRKH-AS1 in Huh7 cells after TDRKH-AS1 knockdown. TDRKH-AS1 was successfully restored after it was knocked down in Huh7 cells(A). Restoration of TDRKH-AS1 could restore the cell growth of Huh7 cells(B).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bu, X., Ma, L., Liu, S. et al. A novel qualitative signature based on lncRNA pairs for prognosis prediction in hepatocellular carcinoma. Cancer Cell Int 22, 95 (2022). https://doi.org/10.1186/s12935-022-02507-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-022-02507-z