
Abstract
Background
Accumulated evidences have demonstrated that long non-coding RNAs (lncRNAs) are correlated with prognosis of patients with hepatocellular carcinoma. The current study aimed to develop and validate a prognostic lncRNA signature to improve the prediction of overall survival in hepatocellular carcinoma patients.
Methods
The study cohort involved 348 hepatocellular carcinoma patients with lncRNA expression information and overall survival information. Through gene mining approach, the current study established a prognostic lncRNA signature (named LncRNA risk prediction score) for predicting the overall survival of hepatocellular carcinoma patients.
Results
The current study built a predictive nomogram based on ten prognostic lncRNA predictors through Cox regression analysis. In model group, the Harrell’s concordance indexes of LncRNA risk prediction score were 0.811 (95% CI 0.769–0.853) for 1-year overall survival, 0.814 (95% CI 0.772–0.856) for 3-year overall survival and 0.796 (95% CI 0.754–0.838) for 5-year overall survival respectively. In validation cohort, the Harrell’s concordance indexes of LncRNA risk prediction score were 0.779 (95% CI 0.737–0.821), 0.828 (95% CI 0.786–0.870) and 0.796 (95%CI 0.754–0.838) for 1-year survival, 3-year survival and 5-year survival respectively. LncRNA risk prediction score could stratify hepatocellular carcinoma patients into low risk group and high risk group. Further survival curve analysis demonstrated that the overall survival rate of high risk patients was significantly poorer than that of low risk patients (P < 0.001).
Conclusions
In conclusion, the current study developed and validated a prognostic signature to predict the individual mortality risk for hepatocellular carcinoma patients. LncRNA risk prediction score is helpful to identify the patients with high mortality risk and optimize the individualized treatment decision. The web calculator can be used by click the following URL: https://zhangzhiqiao2.shinyapps.io/Smart_cancer_predictive_system_HCC_3/.
Similar content being viewed by others

Background
Hepatocellular carcinoma (HCC), as a serious public health problem, is the sixth most common malignant tumor and ranks second in the causes of cancer related death [1]. Since HCC patients at early stage usually had no obvious symptoms, most HCC patients were diagnosed at advanced stage. Despite the great advances in terms of early diagnosis and clinical therapy, the overall survival (OS) of HCC patients remains unsatisfactory [2]. It has been reported that the actual 10-year survival rate was merely 7.2% after surgical resection through a meta analysis with 4197 HCC patients [3]. Therefore, a reliable prognostic signature is needed to monitor HCC patients with poor prognosis and subsequently optimize the clinical treatment decision.
Long non-coding RNAs (lncRNAs), as a class of RNAs > 200 nucleotides in length, may act important roles in biological processes [4, 5]. Several lncRNAs have been reported to be correlated with survival of HCC patients [6, 7]. Recently, several prognostic signatures based on lncRNA expression data have been built to predict the prognosis of HCC patients [8,9,10]. However, these were several limitations for clinical application of these previous prognostic signatures. Firstly, these prognostic signatures provided only simple scores of overall survival but not percentages of individual mortality risk. Secondly, it is too difficult to calculate the risk scores through these complicated prognostic signatures. Meanwhile, the difference and influence of different gene detection platforms and different transformation methods of original gene expression values should be taken into account for clinical application of these prognostic signatures.
Therefore, the present study aimed to build and validate a prognostic model to predict the prognosis of HCC patients using lncRNA expression data downloaded from The Cancer Genome Atlas (TCGA) database. The present study was carried out in accordance with the suggestions by Transparent Reporting of a multivariable prediction model for Individual Prognosis Or Diagnosis (TRIPOD) [11].
Materials and methods
Protocol approval
The present study downloaded the original study dataset from The Cancer Genome Atlas (TCGA) database. The download and analysis of the study dataset strictly adhered to the relevant data policies of TCGA database.
The gene expression dataset
The gene expression dataset was downloaded from TCGA database (January 28, 2018, https://tcga-data.nci.nih.gov/docs/publications/tcga/). The original gene expression data were generated on illumina HiSeq 2000 RNA Sequencing platform. The download gene expression dataset involved 371 hepatocellular carcinoma samples and 50 normal samples with 60,488 original gene expression values. The lncRNAs descripted in GENCODE Resource database (release 27, mapped to GRCh37, https://www.gencodegenes.org/) were selected for further study. There were 14,449 lncRNAs included in the present study for further analysis.
Differential expression analysis
The lncRNAs which original expression values < 1 were filtered out from the present study. Then the lncRNA expression values were further standardized through method of Trimmed Mean of M [12]. The criteria of differential gene selection were P value < 0.05 and |log2fold change| > 2.
Clinical dataset
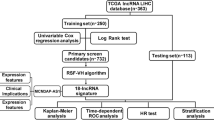
There were 376 HCC patients in the clinical dataset from TCGA database. The study endpoint in the current study was overall survival. To avoid the effects of unrelated confounding factors, 20 HCC patients with overall survival less than 1 month were excluded from the present study. Eight patients without lncRNA expression information were excluded from the present study. Finally, there were 348 HCC patients enrolled the final survival analysis (Fig. 1). The study period of The Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA-LIHC) cohort was from 2010 to 2015. The maximum value and the minimum value of the overall survival time were 120.7 months and 1.0 month. The missing data were recorded as “NA” in the present study. The mean ± standard deviation of age of HCC patients was 59.5 ± 13.4 years in model group. The mean ± standard deviation of follow-up period was 840 ± 701 days. There were 130 (37.4%) out of 348 HCC patients died in the follow-up period.

The flowchart in the current study. TCGA The Cancer Genome Atlas
Internal validation
We carried out an internal validation to validate the predictive performance of the present prognostic model. The validation dataset was constructed by drawing 348 HCC patients using bootstrap resampling method, which was recommended for internal validation of prognostic model [13, 14].
Statistical analysis
Continuous variables in the present study were presented as mean ± standard deviation (SD). The t-test or Mann–Whitney U test was performed to compare the differences of continuous variables as appropriate. The Chi-squared test or Fisher’s exact test was performed to compare the differences of categorical variables as appropriate. Time-dependent receiver operating characteristic (ROC) curves and Harrell’s concordance index (C-index) were performed to assess the predictive accuracy of prognostic models. The statistical analyses were carried out by using SPSS Statistics 19.0 (SPSS Inc., an IBM Company) and R software (version 3.4.4). The following R packages, such as “pROC”, “plyr”, “rms”, “survival”, “timeROC “ and “glmnet “, were performed as appropriate in the present study. P < 0.05 was defined as the criteria of statistical significance.
Results
Study group
Three hundred and forty-eight HCC patients were eventually included in the final survival analysis. The average age of 348 HCC patients was 59.5 ± 13.4 years and the average overall survival time of 348 HCC patients was 28.0 ± 23.7 months in the current study. One hundred and thirty (37.4%) patients out of 348 HCC patients died within the follow-up period in model group. The comparisons of basic characteristics between model group (Additional file 1) and validation cohort (Additional file 2) were summarized in Table 1. There were no significant differences in terms of basic characteristics between model group and validation cohort.
Differential expression analysis
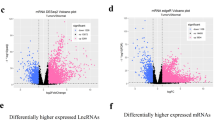
The differential expression analysis between 371 cancer samples and 50 normal samples was performed by using “edgeR” package. Through “edgeR” package, one thousand and five lncRNAs were identified for further survival analysis. The heat map was presented in Additional file 3: Figure S1 and volcano map was presented in Additional file 4: Figure S2.
Construction of prognostic nomogram
The univariate Cox regression analyses were conducted to screen the potential lncRNA predictors for overall survival of HCC patients. Based on the potential lncRNA candidates identified by univariate Cox regression analyses, ten lncRNA predictors for overall survival were finally ascertained through multivariate Cox regression analysis. The relevant model information of ten lncRNA candidates were presented in Table 2. The median values of lncRNA expression values were used as cut-off values to transform the original lncRNA expression values into “1” (as high expression) and “0” (as low expression).
Therefore, a prognostic nomogram (Fig. 2) was built by using the expression values of ten lncRNA predictors: LncRNA risk prediction score = (LINC01559 * 0.771) + (MYLK_AS1 * 0.528) + (RP11_150012.3 * 0.728) − (RP11_92C4.6 * 0.509) − (RASGRF2_AS1 * 0.765) + (LINC01116 * 0.731) + (C2orf48 * 0.563) + (LINC00856 * 0.418) + (LINC02003 * 0.483) + (RP11_363N22.3 * 0.432).

The LncRNA risk prediction score for prediction of overall survival in hepatocellular carcinoma patients
Predictive performance of LncRNA risk prediction score
Through the median value of LncRNA risk prediction score, 348 patients in model group were stratified into low risk group (n = 174) and high risk group (n = 174). As shown in Fig. 3a, the overall survival rate of low risk patients was significantly higher than that of high risk patients (P < 0.001). The distribution of LncRNA risk prediction score was presented in Fig. 3b. The overall survival status and overall survival time were presented in Fig. 3c. The Harrell’s concordance index (C-index) of LncRNA risk prediction score was 0.761 (95% CI 0.719–0.803) for overall survival in model group.

The survival curves of hepatocellular carcinoma patients in model group (a). The distribution of LncRNA risk prediction score (b), survival status and survival time (c) in model group
Clinical application of LncRNA risk prediction score
Time-dependent receiver operating characteristic curves were drawn to depict the clinical application of LncRNA risk prediction score for OS. The C-indexes of LncRNA risk prediction score were 0.811 (95% CI 0.769–0.853) for 1-year overall survival, 0.814 (95% CI 0.772–0.856) for 3-year overall survival and 0.796 (95% CI 0.754–0.838) for 5-year overall survival respectively (Fig. 4a). There were good agreements between predictive survival probability and actual overall survival percentage in calibration curves for 1-year survival (Fig. 4b), 3-year survival (Fig. 4c) and 5-year survival (Fig. 4d).

Performance of LncRNA risk prediction score in model group: time-dependent receiver operating characteristic curves (a); calibration curve for 1-year overall survival (b); calibration curve for 3-year overall survival (c); calibration curve for 5-year overall survival (d)
Internal validation of LncRNA risk prediction score
A internal validation cohort (n = 348) was drawn by random drawing with replacement method from model cohort (n = 348). The calculating method of LncRNA risk prediction scores for patients in validation cohort was as same as the previous formula of LncRNA risk prediction score in model cohort. Then 348 HCC patients in validation cohort were stratified into low risk group (n = 174) and high risk group (n = 174) through the previous cut-off value in model cohort. The survival curve analysis (Fig. 5a) indicated that the overall survival rate in high risk group was significantly poorer than that in low risk group (P < 0.001). The distribution of LncRNA risk prediction score was presented in Fig. 5b. The survival status and survival time were presented in Fig. 5c. The C-index of LncRNA risk prediction score was 0.745 (95% CI 0.703–0.787) for OS in validation cohort.

The survival curves of hepatocellular carcinoma patients in validation cohort (a). The distribution of LncRNA risk prediction score (b), survival status and survival time (c) in validation cohort
Clinical application of LncRNA risk prediction score in validation cohort
In validation cohort, the C-indexes of LncRNA risk prediction score were 0.779 (95% CI 0.737–0.821), 0.828 (95% CI 0.786–0.870) and 0.796 (95% CI 0.754–0.838) for 1-year survival, 3-year survival and 5-year survival respectively (Fig. 6a). There were good agreements between predictive survival probability and actual overall survival percentage in calibration curves for 1-year survival (Fig. 6b), 3-year survival (Fig. 6c) and 5-year survival (Fig. 6d).

Performance of LncRNA risk prediction score in validation cohort: time-dependent receiver operating characteristic curves (a); calibration curve for 1-year overall survival (b); calibration curve for 3-year overall survival (c); calibration curve for 5-year overall survival (d)
Independence assessment of LncRNA risk prediction score
Multivariate Cox regression analyses were carried out to explore the independence of LncRNA risk prediction score for OS of HCC patients. The pathological diagnosis was carried out in accordance with the suggestions of the American Joint Committee on Cancer (AJCC). After adjusting the confounding effects of pathological parameters, gender and age, multivariate Cox regression analyses indicated that LncRNA risk prediction score was an independent influence factor for OS of HCC patients (Table 3).
Survival curve analysis of ten lncRNAs in LncRNA risk prediction score
The survival curve analysis of lncRNAs in LncRNA risk prediction score was present in Fig. 7. As shown in Fig. 7, OS was significantly different according to ten lncRNAs in LncRNA risk prediction score (P < 0.001).

The survival curves of ten lncRNAs in LncRNA risk prediction score
Pathological stage subgroup analysis
Pathological stage was an important influence factor for overall survival of HCC patients. As shown in Fig. 8, OS in high risk group was significantly poorer than that in low risk group in different pathological stages, indicating that the predictive performance of LncRNA risk prediction score for OS was stable and reliable in different pathological stage subgroups.

Pathological stage subgroup analysis
Functional enrichment analysis
According to the criteria of P value < 0.05 and |Spearman correlation coefficient| > 0.7, 162 mRNA genes were significantly co-expressed with prognostic lncRNAs included in LncRNA risk prediction score. Functional enrichment analysis was performed through the Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov/). Gene ontology (GO) biological process enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathway analysis were presented in Fig. 9. Functional enrichment analysis indicated that the co-expressed genes were mainly enriched in mitotic nuclear division, cell division, DNA replication, DNA repair, regulation of cell cycle, DNA-dependent ATPase activity, and ATPase activity.

Functional enrichment analysis of prognostic signature: a biological process; b molecular function; c cellular component; d KEGG pathway. KEGG Kyoto Encyclopedia of Genes and Genomes
Ten-group risk stratification chart
To explore the predictive performance of LncRNA risk prediction score for OS, a 10-group risk stratification chart was presented in Fig. 10 for model cohort. The discriminative ability of LncRNA risk prediction score for 1 year, 2 year, and 3 year OS were showed in Fig. 10a–c.

Ten-group risk stratification chart: a for 1-year overall survival; b for 2-year overall survival; c for 3-year overall survival
Association between prognostic lncRNAs and tumors of digestive system
We further explored the association between prognostic lncRNAs and tumors of digestive system through MNDR v2.0 database (http://www.rna-society.org/mndr/index.html). MNDR v2.0 database integrated clinical evidences from 14 resources and provided a confidence score for each ncRNA-disease association.
RASGRF2_AS1, LINC00856 and LINC01116 were related with hepatocellular carcinoma (score 0.1097), stomach cancer (score 0.1097), and colorectal cancer (score 0.1097). MYLK_AS1 was related with stomach cancer (score 0.8473). RP11_150O12.3 was related with stomach cancer (score 0.4752). LINC01559 and C2orf48 were related with stomach cancer (score 0.1097).
Discussion
The current study developed and validated a prognostic model named LncRNA risk prediction score, which was helpful to predict the individual mortality risk and identify the patients with high mortality risk. LncRNA risk prediction score could help HCC patients with high mortality risk optimize their individualized clinical decision.
LncRNA risk prediction score, as a prognostic nomogram, provided a noninvasive preoperative predictive method for overall survival of HCC patients. The nomogram predictive chart has been used as predictive tool for prediction of prognosis in different cancers [15, 16]. The present study constructed LncRNA risk prediction score for OS was based on the following points to consider: First, there is an urgent need for clinical practice to construct a preoperative predictive method to forecast the overall survival of HCC patients before further surgery. The HCC patients with high mortality risk identified by prognostic models would be more willing to accept active treatment such as surgical treatment. Second, for HCC patients without pathological diagnosis information, LncRNA risk prediction score could provide an alternative noninvasive predictive method for overall survival.
The previous prognostic models didn’t present in the current study for the following causes [8,9,10]. First, these prognostic models were developed based on lncRNA expression values generated on different gene detection platforms. Due to the differences between different gene detection platforms, these prognostic models couldn’t be calculated directly in the current study. Second, the previous studies further standardized the original lncRNA expression counts by using different standardization methods. The standardization methods in these previous studies reduced the repeatability and clinical applicability of these prognostic models.
The current study has the following advantages in predicting the overall survival of HCC patients: First, LncRNA risk prediction score, as a simple predictive nomogram, was easy to calculate and understand by patients. Second, the individual mortality risk was presented as percentage of mortality risk, which was easy to interpret the clinical significance of the predictive result for patients without medical knowledge. Third, since this prognostic nomogram didn’t contain pathological parameters, LncRNA risk prediction score was a noninvasive predictive method and subsequently more suitable for preoperative prediction for OS.
There were several shortcomings in the current study. First, LncRNA risk prediction score has not been validated through external study dataset. Therefore it was necessary to validate the predictive performance of LncRNA risk prediction score in different external study population. Second, the sample size of the current study was relevant small and then large prospective multicenter studies are needed to further validate the clinical value of LncRNA risk prediction score for overall survival of HCC patients. Third, the results in the present study depended on gene mining approach and lacked evidences from clinical trials. It is necessary to carry out further clinical research to verify the results in the present study.
Conclusion
In conclusion, the current study developed and validated a prognostic model to predict the individual mortality risk of HCC patients. The LncRNA risk prediction score is helpful to identify the patients with high mortality risk and subsequently optimize the individualized treatment decision.
Availability of data and materials
The datasets analyzed in the current study are provided as the additional documents in the end of the current article. Smart Cancer Predictive System tools were designed by Zhiqiao Zhang from Precision Medical Development Group of Institute of Hepatology, Shunde Hospital, Southern Medical University. The following calculator is the fifth predictive tool in Smart Cancer Predictive System. The web calculator can be used by click the following URL: https://zhangzhiqiao2.shinyapps.io/Smart_cancer_predictive_system_HCC_3/.
Abbreviations
- HCC:
-
hepatocellular carcinoma
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
the Gene Expression Omnibus
- ROC:
-
receiver operating characteristic
- OS:
-
overall survival
- lncRNA:
-
long non-coding RNA
- miRNA:
-
microRNAs
- HR:
-
hazard ratio
- CI:
-
confidence interval
- AJCC:
-
the American Joint Committee on Cancer
- SD:
-
standard deviation
- DCA:
-
decision curve analysis
- ceRNA:
-
competitive endogenous RNA
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
Chapman WC, Klintmalm G, Hemming A, Vachharajani N, Majella Doyle MB, DeMatteo R, Zaydfudim V, Chung H, Cavaness K, Goldstein R, et al. Surgical treatment of hepatocellular carcinoma in North America: can hepatic resection still be justified? J Am Coll Surg. 2015;220(4):628–37.
Gluer AM, Cocco N, Laurence JM, Johnston ES, Hollands MJ, Pleass HC, Richardson AJ, Lam VW. Systematic review of actual 10-year survival following resection for hepatocellular carcinoma. HPB. 2012;14(5):285–90.
Zhu XT, Yuan JH, Zhu TT, Li YY, Cheng XY. Long noncoding RNA glypican 3 (GPC3) antisense transcript 1 promotes hepatocellular carcinoma progression via epigenetically activating GPC3. FEBS J. 2016;283(20):3739–54.
Jiang X, Liu W. Long noncoding RNA highly upregulated in liver cancer activates p53-p21 Pathway and promotes nasopharyngeal carcinoma cell growth. DNA Cell Biol. 2017;36(7):596–602.
Li T, Xie J, Shen C, Cheng D, Shi Y, Wu Z, Deng X, Chen H, Shen B, Peng C, et al. Upregulation of long noncoding RNA ZEB1-AS1 promotes tumor metastasis and predicts poor prognosis in hepatocellular carcinoma. Oncogene. 2016;35(12):1575–84.
Zhang JY, Weng MZ, Song FB, Xu YG, Liu Q, Wu JY, Qin J, Jin T, Xu JM. Long noncoding RNA AFAP1-AS1 indicates a poor prognosis of hepatocellular carcinoma and promotes cell proliferation and invasion via upregulation of the RhoA/Rac2 signaling. Int J Oncol. 2016;48(4):1590–8.
Ma Y, Luo T, Dong D, Wu X, Wang Y. Characterization of long non-coding RNAs to reveal potential prognostic biomarkers in hepatocellular carcinoma. Gene. 2018;663:148–56.
Sui J, Miao Y, Han J, Nan H, Shen B, Zhang X, Zhang Y, Wu Y, Wu W, Liu T, et al. Systematic analyses of a novel lncRNA-associated signature as the prognostic biomarker for Hepatocellular Carcinoma. Cancer Med. 2018;7(7):3240–56.
Wang Z, Wu Q, Feng S, Zhao Y, Tao C. Identification of four prognostic LncRNAs for survival prediction of patients with hepatocellular carcinoma. PeerJ. 2017;5:e3575.
Collins GS, Reitsma JB, Altman DG, Moons KG, Group T. Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD): the TRIPOD statement. The TRIPOD Group. Circulation. 2015;131(2):211–9.
Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25.
Blackstone EH. Breaking down barriers: helpful breakthrough statistical methods you need to understand better. J Thorac Cardiovasc Surg. 2001;122(3):430–9.
Grunkemeier GL, Wu Y. Bootstrap resampling methods: something for nothing? Ann Thorac Surg. 2004;77(4):1142–4.
Li Y, Xia Y, Li J, Wu D, Wan X, Wang K, Wu M, Liu J, Lau WY, Shen F. Prognostic nomograms for pre- and postoperative predictions of long-term survival for patients who underwent liver resection for huge hepatocellular carcinoma. J Am Coll Surg. 2015;221(5):962–974.e964.
Tian X, Zhu X, Yan T, Yu C, Shen C, Hong J, Chen H, Fang JY. Differentially expressed lncRNAs in gastric cancer patients: a potential biomarker for gastric cancer prognosis. J Cancer. 2017;8(13):2575–86.
Acknowledgements
We appreciated the Cancer Genome Atlas database, the Gene Expression Omnibus database, and the cBioPortal database. The idea of web calculator in this article was inspired by QCancer® tools designed by Mr Gary S Collins and his group, and we would like to express our sincere thanks to Mr Gary S Collins and his group. We sincerely thanked Qingmei Liu, a computer professional programmer, for her support on program coding and software development.
Funding
This study was supported by Guangdong Provincial Health Department and Guangdong Provincial Financial Department. The Grant numbers were: B2018237 (Grant recipient: Zhiqiao Zhang) and A2016450 (Grant recipient: Zhiqiao Zhang). The total capital was RMB 15000. The funders had no role in study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
ZQZ, and PW designed the current study; ZQZ, JL, TSH, YLOY, YYH, and QBL collected and analyzed the study data; ZQZ, JL, TSH, and PW wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We downloaded and analyzed the original study dataset in accordance with the data relevant policies of The Cancer Genome Atlas (TCGA) database and therefore no additional ethics approval was needed.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1.
Model cohort dataset.
Additional file 2.
Validation cohort dataset.
Additional file 3: Figure S1.
Heat map.
Additional file 4: Figure S2.
Volcano map.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, Z., Ouyang, Y., Huang, Y. et al. Comprehensive bioinformatics analysis reveals potential lncRNA biomarkers for overall survival in patients with hepatocellular carcinoma: an on-line individual risk calculator based on TCGA cohort. Cancer Cell Int 19, 174 (2019). https://doi.org/10.1186/s12935-019-0890-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-019-0890-2