
Abstract
Background
Saccharomyces cerevisiae generally consumes glucose to produce ethanol accompanied by the main by-products of glycerol, acetic acid, and lactic acid. The minimization of the formation of by-products in S. cerevisiae was an effective way to improve the economic viability of the bioethanol industry. In this study, S. cerevisiae GPD2, FPS1, ADH2, and DLD3 genes were knocked out by the Clustered Regularly Interspaced Short Palindromic Repeats Cas9 (CRISPR-Cas9) approach. The mechanism of gene deletion affecting ethanol metabolism was further elucidated based on metabolic flux and transcriptomics approaches.
Results
The engineered S. cerevisiae with gene deletion of GPD2, FPS1, ADH2, and DLD3 was constructed by the CRISPR-Cas9 approach. The ethanol content of engineered S. cerevisiae GPD2 Delta FPS1 Delta ADH2 Delta DLD3 Delta increased by 18.58% with the decrease of glycerol, acetic acid, and lactic acid contents by 22.32, 8.87, and 16.82%, respectively. The metabolic flux analysis indicated that the carbon flux rethanol in engineered strain increased from 60.969 to 63.379. The sequencing-based RNA-Seq transcriptomics represented 472 differential expression genes (DEGs) were identified in engineered S. cerevisiae, in which 195 and 277 genes were significantly up-regulated and down-regulated, respectively. The enriched pathways of up-regulated genes were mainly involved in the energy metabolism of carbohydrates, while the down-regulated genes were mainly enriched in acid metabolic pathways.
Conclusions
The yield of ethanol in engineered S. cerevisiae increased with the decrease of the by-products including glycerol, acetic acid, and lactic acid. The deletion of genes GPD2, FPS1, ADH2, and DLD3 resulted in the redirection of carbon flux.
Similar content being viewed by others
Background
Ethanol mainly produced by Saccharomyces cerevisiae is widely applied in the chemical industry, beverages, bioethanol, pharmaceuticals, and cosmetics [1]. 90–95% of ethanol is produced via the anaerobic fermentation approach [2]. Ethanol fermentation biochemistry includes substrate degradation pathways (glycolysis, alcoholic and glyceropyruvic fermentation, xylose catabolic pathways, and glycerol assimilation) and metabolic regulation pathways between fermentation and respiration (Pasteur effect, Kluyver effect, Crabtree effect, and Custers effect) [3,4,5,6]. In the process of carbon flow metabolism in S. cerevisiae, 5% of carbon source is converted into glycerol as a byproduct [7]. Although glycerol plays a physiological role in osmoregulation and regulating redox balance, the excessive formation of glycerol will reduce the utilization rate of sugar, and then affect the production rate of ethanol [8].
The glycerol formation can be effectively regulated by knocking out the glycerol formation pathway, preventing glycerol secretion, and regulating redox balance. Therefore, the redirection of carbon flow and the influence of the intracellular redox potential pathway will affect glycerol formation and ethanol production. Glycerol is produced from dihydroxyacetone phosphate (DHAP) in the presence of glycerol-3-phosphate dehydrogenase and glycerol-3-phosphatase phosphatase. NAD+ dependent glycerol-3-phosphate dehydrogenase has two isoforms of Gpd1 and Gpd2 in S. cerevisiae, which catalyze the reduction of DHAP to glycerol-3-phosphate by synergetic catalysis approach [9, 10]. Redox cofactors play a role in cellular metabolism by participating in numerous biochemical reactions [11]. The maintenance of redox balance is a fundamental requirement for cellular metabolism and cell growth [12]. Thus, GPD2 affects the carbon metabolic pathway of glycerol formation by regulating redox potential.
In S. cerevisiae, ethanol is produced from pyruvate via the following two steps: decarboxylation of pyruvate to acetaldehyde by pyruvate decarboxylase; acetaldehyde is further reduced to ethanol by alcohol dehydrogenase (Adh). ADH2 encoding for alcohol dehydrogenase II catalyzes ethanol oxidation toward acetaldehyde [13]. Thus, during the pathway engineering, less ethanol is consumed by the disruption of alcohol dehydrogenase 2 (Adh2) activity [14]. In addition, ADH2 deletion is involved in the increased demand for NAD+ regeneration in the carbon metabolism. Both cofactor removal and consumption reduction could result in the metabolic cofactor imbalance [15, 16]. Glycerol is mainly exported across the plasma membrane in S. cerevisiae through the protein channel Fps1 regulated by extracellular osmolarity [17, 18]. Fps1 is a member of the major intrinsic protein (MIP) family as channel proteins, which contain six transmembrane domains [19]. S. cerevisiae fps1Δ mutant exhibits the intracellular accumulation of glycerol. The glycerol accumulation triggers other regulatory systems for the reduction of glycerol biosynthesis, which then results in an increase of ethanol yield [20]. In addition, d-lactate dehydrogenase 3 (Dld3) in S. cerevisiae is involved in the conversion of d-lactate to pyruvate [21].
To improve the ethanol yield and decrease the formation of by-products, Clustered Regularly Interspaced Short Palindromic Repeats Cas9 (CRISPR-Cas9) approach was used to knock out S. cerevisiae GPD2, FPS1, ADH2, and DLD3 to modify the metabolic pathway in this study (Fig. 1). The fermentation characteristics of the engineered S. cerevisiae strain were investigated to analyze the effect of gene deletion on the production of ethanol and by-products. In addition, the molecular mechanism was comprehensively elucidated based on the metabolic flux and transcriptomics approaches.

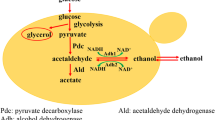
Modification of carbon metabolism pathway of S. cerevisiae for improvement of ethanol yield in this study. G-6-P: glucose; F-6-P: fructose-6-phosphate; F-1,6-BP: Fructose 1,6-bisphosphate; DHAP: dihydroxyacetone phosphate; GL3P: glycerol-3-phosphate; DHA: dihydroxyacetone; G-3-P: glyceraldehyde-3-phosphate; PEP: phosphoenolpyruvic acid; PYR: pyruvic acid; α-KET: α-ketoglutarate; Suc: Succinic acid; OAA: Oxaloacetic acid
Results
Effect of gene deletion on the grow of engineered S. cerevisiae SCGFAD
Four genes of GPD2, FPS1, ADH2, and DLD3 in S. cerevisiae were successively knocked out by the CRISPR-Cas9 approach. Each deletion of the gene was confirmed by PCR and sequencing identification. The S. cerevisiae mutant with GPD2, FPS1, ADH2, and DLD3 deletion was named S. cerevisiae SCGFAD. S. cerevisiae was inoculated into YPD liquid medium containing 50 g/L glucose. The growth of S. cerevisiae was determined by measuring the absorbance at the wavelength of 600 nm (Fig. 2). The result showed that S. cerevisiae SCGFAD possessed a similar growth curve to the wild-type strain. During the fermentation of 0–24 h, the growth curve of S. cerevisiae SCGFAD mutant was consistent with that of the wild-type strain. During fermentation of 24–72 h, the absorbance of S. cerevisiae SCGFAD at the wavelength of 600 nm was slightly below that of the wild-type strain. The OD 600 nm values of engineered S. cerevisiae SCGFAD and wild-type strain were 9.45 and 9.83 after fermentation for 72 h, respectively. Thus, the deletion of four genes of GPD2, FPS1, ADH2, and DLD3 did not markedly affect the growth of engineered S. cerevisiae SCGFAD.

Effect of gene deletion on the growth of engineered S. cerevisiae SCGFAD
Ethanol production and glucose consumption of S. cerevisiae SCGFAD
The ethanol production and glucose consumption in S. cerevisiae SCGFAD during fermentation were investigated in comparison with the wild-type strain. The results showed that the residual content of glucose from S. cerevisiae SCGFAD was similar to that from the wild-type strain. The initial glucose content of 50 g/L decreased to 0 after fermentation for 48 h (Fig. 3). During the process of glucose consumption, the ethanol content of S. cerevisiae SCGFAD increased simultaneously. During fermentation of 0–24 h, both S. cerevisiae SCGFAD and wild-type strain exhibited a similar ethanol production trend. However, during fermentation of 24–72 h, the ethanol content from S. cerevisiae SCGFAD was higher than that from the wild-type strain. S. cerevisiae SCGFAD possessed an ethanol content of 23.29 g/L, which was 1.19 folds of the wild-type strain (19.64 g/L). Thus, S. cerevisiae SCGFAD with GPD2, FPS1, ADH2, and DLD3 deletion exhibited a higher ethanol production rate compared with the wild-type strain by modifying the metabolic pathway.

Ethanol production and glucose consumption of the wild-type strain and S. cerevisiae SCGFAD during fermentation
Production of glycerol, acetic acid, and lactic acid in S. cerevisiae SCGFAD
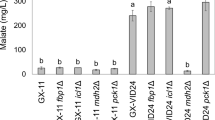
Glycerol, acetic acid, and lactic acid were the by-products of S. cerevisiae during the fermentation for ethanol production. In this study, the contents of glycerol, acetic acid, and lactic acid during fermentation were determined to investigate the effect of gene deletion on the by-product formation (Fig. 4). During fermentation, the glycerol content in the cultivation medium from S. cerevisiae SCGFAD was lower than that of wild-type S. cerevisiae (Fig. 4A). The content of glycerol in broth increased slowly during fermentation of 0–72 h. After fermentation for 72 h, the glycerol content from S. cerevisiae SCGFAD was 1747 mg/L, which was 0.78-fold compared with that from the wild-type S. cerevisiae (2249 mg/L).

Production of glycerol, acetic acid, and lactic acid in the wild-type strain and S. cerevisiae SCGFAD during fermentation. A Glycerol content of the wild-type strain and S. cerevisiae SCGFAD; B the contents of acetic acid, and lactic acid of the wild-type strain and S. cerevisiae SCGFAD
The effect of gene deletion in S. cerevisiae SCGFAD on acetic acid and lactic acid content was also investigated compared with the wild-type strain (Fig. 4B). The contents of acetic acid and lactic acid produced by S. cerevisiae SCGFAD were lower than that of wild-type S. cerevisiae. After fermentation of 72 h, the content of acetic acid in the fermentation broth from S. cerevisiae SCGFAD was 113 mg/L, which was 8.87% lower than that of wild-type S. cerevisiae (124 mg/L). In addition, after fermentation for 72 h, the lactic acid content in the fermentation broth from S. cerevisiae SCGFAD was 6.28 mg/L, which was 16.82% lower than that of wild-type S. cerevisiae (7.55 mg/L). These results indicated that the deletion of the four genes of GPD2, FPS1, ADH2, and DLD3 resulted in the decrease of glycerol, acetate, and lactate contents in S. cerevisiae.
Determination of contents and rates of metabolites
According to the fermentation characteristics of S. cerevisiae, metabolites were in the logarithmic stage when the fermentation time reached 18–30 h. Three-time points of 18 h, 24 h, and 30 h were selected as sampling points to measure the concentrations of glucose, ethanol, glycerol, lactic acid, acetic acid, succinic acid, and biomass. Metabolites and their contents from the wild-type and engineered S. cerevisiae under three different fermentation stages were recorded in Table 1.
Stoichiometric model
According to the metabolic flux model and the chemical reaction equations of the metabolites (Additional file 1), the rate equations of the intermediate metabolites were obtained (Table 2). Total of 15 reactions were used to represent the metabolites in S. cerevisiae. Based on the hypothesis principle of no change in the composition of S. cerevisiae cells during fermentation, the demand coefficients of cell precursor in a unit of mmol/g DCW (dry cell weight) were rm1 = 2.515 rm, rm2 = 0.606 rm, rm3 = 0.601 rm, rm4 = 0.007 rm, rm5 = 0.528 rm, rm6 = 1.756 rm, rm7 = 0.876 rm, rm8 = 1.159 rm, rm9 = 0.834 rm, rmCO2 = 2.610 rm, where rm represented the increase rate of cell biomass (g/h).
The hypothesis equation for metabolic flux analysis is A × r = 0, where A represented an m × n matrix as stoichiometric coefficients, r represented an m-dimensional column vector containing metabolic reaction rates, m represented the number of intracellular reactions, and n referred to the number of intermediate metabolites. In this study, the matrix equations of wild-type S. cerevisiae and SCGFAD were shown in Fig. 5. On the basis of A × r = 0, the cell precursor demand coefficient and known rate were substituted into the rate equation of intermediate metabolites, and the constant term was moved to the right side of the equation.

Rate equations of intermediate metabolites for the stoichiometric model analysis
Metabolic flux analysis of S. cerevisiae SCGFAD
Each flux data was normalized based on the glucose consumption rate (C·mol/ (L·h)). The metabolic flux distribution of S. cerevisiae SCGFAD was drawn according to the calculation results (Fig. 6). The flux r1(82.8753/92.9681) of the glycolytic pathway was dramatically higher than the flux r2(16.6121/6.5337) of the pentose phosphate pathway, which indicated that glycolysis was the main pathway of glucose carbon metabolism in S. cerevisiae. Metabolic flux distribution showed that deletion of GPD2 for glycerol synthesis and FPS1 for transport of glycerol resulted in a decrease in rglycerol from 3.7300 to 1.9220. The carbon flux r5 for the glycerol synthesis pathway decreased dramatically from 3.8525 to 2.0411. Carbon flux redistribution resulted in carbon flux r4 for the catalysis of dihydroxyacetone phosphate to glyceraldehyde-3-phosphate increased from 42.1628 to 46.2194. The increase of carbon fluxes r6, r7, r8, and r9 for the conversion of glyceraldehyde-3-phosphate to acetaldehyde contributed to reducing glycerol production and increasing ethanol yield. In addition, DLD3 deletion decreased the carbon flux r11 from 0.6211 to 0.6011 in the reaction node of acetaldehyde and acetate. The co-knockout of four genes increased the carbon flux rethanol from 60.969 to 63.379, which caused the increase in ethanol yield.

Metabolic flux distribution of S. cerevisiae SCGFAD. The black numbers on the left of the slash and the red numbers on the right of the slash represented the fluxes of wild-type S. cerevisiae and S. cerevisiae SCGFAD, respectively. 3PG: 3-phosphate-glycerate; AC.: acetaldehyde; AC.A: acetic acid; R5P: ribose-5-phosphate
S. cerevisiae cDNA Library and DEGs analysis
The construction of the S. cerevisiae mRNA Library was performed by the following steps including accurate quantification of total RNA, mRNA purification and fragmentation, synthesis and purification of the double-stranded cDNA, end repair and dA tail addition, ligation, purification of ligation products, fragment size sorting, and library amplification. After Illumina Hiseq™ sequencing, the data were used to perform the analysis of gene expression differences. A total of 472 DEGs from S. cerevisiae SCGFAD were identified compared with wild-type S. cerevisiae (Additional file 2), in which 195 and 277 genes were significantly up-regulated and down-regulated, respectively (Fig. 7).

DEGs analysis of S. cerevisiae SCGFAD
GO functional annotation of differential genes
The results of GO analysis showed that 472 DEGs were classified into three broad categories of biological processes, cellular components, and molecular function (Fig. 8). There were 19 GO terms with up- and down-regulated genes in biological processes. DEGs were mainly concentrated in the cellular component organization or biogenesis, cellular processes, and metabolic processes. There were 13 GO terms involved in the up-regulated and down-regulated genes in the cellular components. The top three categories of cells, cellular parts, and organelles accounted for the most of DEGs. In addition, 11 of GO terms were involved in the up- and down-regulated genes in the molecular function. DEGs were enriched in terms of binding and catalytic activity. In the GO analysis, the most clearly enriched term for up-regulated genes was GO:0005488 defined as binding in molecular function. The main up-regulated genes of COX1, CYC7, and HXK1 were involved in mitochondrial electron transport and glycolysis pathways, respectively. The result indicated the gene deletion resulted in the increase of electron transfer activity and glucose decomposition in S. cerevisiae SCGFAD.

GO functional annotation of S. cerevisiae SCGFAD
KEGG enrichment analysis
KEGG databases were used to determine whether DEGs were involved in specific pathways. The enrichment analysis of up-regulated genes included the top 30 enriched pathways among the 98 pathways (Fig. 9). The enriched pathways of up-regulated genes mainly included energy metabolism processes, such as starch and sucrose metabolism, glycolysis/gluconeogenesis, galactose metabolism, fructose, and mannose metabolism, carbohydrate digestion and absorption, and amino acid and nucleotide sugar metabolism. In addition, up-regulated genes were also enriched in signaling pathways, such as P53 signaling pathway, MAPK signaling pathway, insulin signaling pathway, and HIF-1 signaling pathway. The enrichment analysis of down-regulated genes showed only the top 30 from 79 enriched pathways (Fig. 10). The down-regulated genes were mainly enriched in acid metabolic pathways, such as pyruvate metabolism, α-linolenic acid metabolism, glyoxylate and dicarboxylic acid metabolism, and fatty acid metabolism. In addition, the meiosis-yeast and cell cycle-yeast pathways were also enriched for down-regulated genes.

KEGG enrichment analysis for up-regulated genes

KEGG enrichment analysis for down-regulated genes
Discussion
Rational metabolic engineering was used to redirect the metabolic flow in S. cerevisiae by engineering metabolic pathways mainly including enzymes, transporters, and regulatory proteins based on available information [22]. S. cerevisiae used for bioethanol production is classified as GRAS (generally regarded as safe) by the U.S. Food and Drug Administration. However, several by-products of glycerol, yeast biomass, carbon dioxide, and other minor products during yeast fermentation lowered the total ethanol yield. The ethanol yield was increased by 12% by deleting GPD1 and GPD2 to completely abolish yeast’s glycerol formation. However, the growth of S. cerevisiae with the double mutations of GPD1 and GPD2 was severely affected [23]. ADH2 plays a crucial role in the conversion of ethanol to acetaldehyde in S. cerevisiae [24]. The deletion of FPS1 caused the increase of ethanol by 10% [20]. The previous reports also showed the deletion of GPD2 and FPS1 reduced the content of glycerol and organic acids [25, 26]. Due to multiple metabolic pathways involving the conversion of glucose to ethanol, in this study, four genes of GPD2, FPS1, ADH2, and DLD3 were deleted to construct engineered S. cerevisiae with an increase of ethanol yield by 18.58%. In brief, this study provided a genetic modification strategy for the improvement of ethanol yield in S. cerevisiae by regulating the carbon flux distribution and inhibiting the by-product formation on the basis of a comprehensive analysis of ethanol metabolism.
The metabolic flux and sequencing-based RNA-Seq transcriptomics were effective methods to elaborate the molecular mechanism of arabinose fermentation in engineered S. cerevisiae [27]. In this study, metabolic flux analysis showed the deletion of four genes resulted in the increase of carbon flux rethanol from 60.969 to 63.379, which meant more carbon flux in ethanol production in engineered S. cerevisiae. KEGG enrichment analysis showed the enriched pathways of up-regulated genes were mainly involved in the energy metabolism of carbohydrates, while the down-regulated genes were mainly enriched in acid metabolic pathways. The transcriptomics analysis provided large information on gene expression and function differences in engineered S. cerevisiae due to gene deletion.
In this study, the lactic acid contents of the wild-type strain, S. cerevisiae GPD2 delta FPS1 delta ADH2 delta mutant, and S. cerevisiae GPD2 delta FPS1 delta ADH2 delta DLD3 delta mutant were 7.55, 6.59, and 6.28 mg/L, respectively. Correspondingly, the ethanol concentrations of the above three strains were 19.64, 23.12, and 23.29 g/L, respectively. The increased amount of ethanol (0.17 g/L) was much higher than the decreased amount of lactic acid (0.31 mg/L) in S. cerevisiae mutant with four-gene deletion compared with three-gene deletion. Thus, DLD3 deletion could result in the redirection of other metabolisms related to ethanol production. This study further indicated that DLD3 deletion caused the decrease of the carbon flux r11 from 0.6211 to 0.6011 in the reaction node of acetaldehyde and acetate. However, the influence degree and regulation mechanism of DLD3 deletion on other carbon metabolism pathways need to be further clarified.
Conclusion
Four genes of S. cerevisiae GPD2, FPS1, ADH2, and DLD3 involving ethanol production were knocked out by the CRISPR-Cas9 approach. The gene deletion in engineered S. cerevisiae caused the increase of ethanol content by 18.58%. In addition, the contents of glycerol, acetic acid, and lactic acid decreased by 22.32, 8.87, and 16.82%, respectively. The carbon flux rethanol in engineered strain increased from 60.969 to 63.379, which represented more carbon flux in ethanol production. In addition, 472 DEGs from S. cerevisiae SCGFAD were identified, in which 195 and 277 genes were significantly up-regulated and down-regulated, respectively. KEGG enrichment analysis indicated the enriched pathways of up-regulated genes mainly were energy metabolism processes, and amino acid and nucleotide sugar metabolism, while the down-regulated genes were mainly enriched in acid metabolic pathways. The engineered S. cerevisiae strain would be applied in bioethanol industry for high-level ethanol production with less formation of by-products.
Materials and methods
Strains, plasmids, primers and culture conditions
The wild-type strain used for gene knockout in this study was S. cerevisiae S288c. S. cerevisiae GPD2 Delta FPS1 Delta ADH2 Delta DLD3 Delta mutant (SCGFAD) was constructed by knocking out GPD2, FPS1, ADH2, and DLD3 genes of wild-type S. cerevisiae. Cas9-NTC and gRNA-trp-HYB were from Addgene Company (Watertown, MA, USA). GPD2-gRNA, FPS1-gRNA, ADH2-gRNA, and DLD3-gRNA plasmids carrying hygromycin B (HyB) resistance gene were amplified from gRNA-trp-HYB with the prepared primers. These plasmids expressing 20-bp gRNA were used to recognize the target gene loci of S. cerevisiae. The four pairs of primers were designed using the Weblink http://chopchop.cbu.uib.no/ online search system for 20 bp of gRNA sequences (Table 3). Four pairs of primers were also designed to amplify the donor DNA of GPD2, FPS1, ADH2, and DLD3. In addition, plasmid Cas9-NTC carrying nuclease and nourseothricin resistance gene was used to cut off the genomic DNA in S. cerevisiae. Yeast extract peptone dextrose medium (YPD) containing 20 g/L of glucose was prepared to culture S. cerevisiae for cell proliferation at 30 °C and 200 rpm. When the OD600nm of S. cerevisiae reached 1, 1 mL of broth was inoculated into a 250-mL conical flask loaded with 100 mL of YPD containing 50 g/L glucose for further fermentation under the conditions of 30 °C and 200 rpm. The solid medium YPDN was prepared by the addition of 80 μg/mL NTC in YPD solid medium to screen the hypothetical transformants with Cas9-NTC integration. In addition, YPDNH medium was prepared by addition of 300 μg/mL HyB and 80 μg/mL NTC in YPD solid medium to screen the hypothetical transformants with the co-integration of Cas9-NTC and gRNA plasmid.
Construction of SCGFAD mutant strain by CRISPR-Cas9 technology
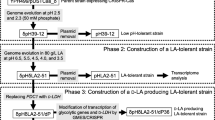
Saccharomyces cerevisiae GPD2, FPS1, ADH2, and DLD3 were sequentially knocked out by the CRISPR-Cas9 technology. The genetically engineered S. cerevisiae with GPD2, FPS1, ADH2, and DLD3 deletion (SCGFAD) was constructed (Fig. 11). The transformation of plasmid in the genome of S. cerevisiae was carried out according to the PEG-mediated LiA-ssDNA method [28]. During the integration of GPD2, Cas9-NTC was transformed into the wild-type S. cerevisiae on the YPDN screening medium at 30 °C. The putative transformants grew on the solid screening medium containing antibiotics after an incubation of 48 h at 30 °C. Then GPD2-gRNA and donor DNA were transformed into the above transformants, and screened on the YPDNH solid medium. After culture for 48 h at 30 °C, the putative colonies were confirmed by PCR to amplify the donor DNA. After sequencing identification, the donor DNA was confirmed to be inserted into the genome DNA. The engineered S. cerevisiae was further used for the other gene integration after the loss of integrated plasmids on the antibiotic-free YPD medium.

Pathway of S. cerevisiae GPD2 Delta FPS1 Delta ADH2 Delta DLD3 Delta mutant construction by the CRISPR-Cas9 approach
Effect of gene knocking out on the growth of S. cerevisiae
The effect of four-gene deletion on the growth and proliferation of SCGFAD mutant was investigated by the measurement of cell concentration. The fermentation broth of 1 mL with cell concentration of 1 OD600nm was inoculated into a 250-mL conical flask containing 100 mL YPD. The fermentation was performed at 30 °C with a shaking speed of 200 rpm. Then, the broth samples of the SCGFAD mutant and wild-type strain were extracted every 6 h to measure the absorbance at 600 nm of wavelength.
Determination of ethanol and by-products contents
High-Performance Liquid Chromatography (HPLC) method was used to measure the contents of ethanol and by-products. The parameters for the measurement of glucose, ethanol, and glycerol contents were a mobile phase of 0.01 mol/L H2SO4, a flow rate of 0.8 mL/min, column temperature of 50 °C with the instruments of Waters 1525 binary HPLC pump, Waters 2410 refractive index detector, and Shodex SH1011 chromatographic column [29]. In addition, the parameters for the content determination of organic acids, mainly including lactic acid, acetic acid, and succinic acid were detection wavelength of 210 nm, a mobile phase A of 10 mM KH2PO4, a mobile phase B of methanol, flow rate of 1.0 mL/min, column temperature of 30 °C with the instruments of Waters Alliance E2695, Waters 2489 UV detector, and Waters XSelect HSS chromatographic column [30]. The concentration of biomass in the medium was measured by the gravimetric method [31].
Metabolic flux models and quasi-steady-state equation calculations
Saccharomyces cerevisiae cerevisiae metabolic network model was constructed according to the method established by the previous report [31]. The selected reaction pathways were glycolysis pathway (EMP), pentose phosphate pathway (PPP), and tricarboxylic acid cycle pathway (TCA). All pathways of the PPP reduce to a single reaction equation under the quasi-steady-state assumption. A simplified metabolic flux model of S. cerevisiae was constructed according to the above requirements. The cell precursor demand coefficients using unit of mmol/g DCW (cell dry weight) were referred to the previous report [32]. The corresponding stoichiometric model was constructed based on the metabolic flux model and reaction equation of each substance. It was assumed that the reaction in the S. cerevisiae cell was in a pseudo-steady state. The unknown intracellular reaction was calculated by the measured metabolite consumption or production rates. The stoichiometric model was calculated based on the carbon balance of the compound with the rate unit of C·mol/ (L·h). All the calculations were performed by Matlab Software [33].
Construction of cDNA library
Construction of the cDNA library was performed via the following steps: (1) RNA extraction, mRNA purification, and double-stranded cDNA synthesis. The double-stranded cDNA was synthesized after mRNA was purified from total RNA. The purified cDNA samples were subjected to end A tail addition and linker ligation reactions [34]; (2) the products from the amplified cDNA library were purified by Hieff NGS™ DNA Selection Beads with 1:1 of Beads and DNA; (3) the cDNA library was sequenced by Illumina Hiseq™ [35].
Transcriptome analysis
The original image data file processed by Illumina Hiseq™ was analyzed by CASAVA (Base Calling). After the conversion into the original sequencing sequence, Trimmomatic was used to remove sequences with N bases, linker sequences in reads, low-quality sequences (q < 20), and then the clean data were obtained [36, 37]. The obtained sequences after quality control were compared with the reference genome (S288c) by HISAT2 [38]. The results were counted by the RSeQC method [39]. According to the sequencing results, Transcripts Per Million (TPM) were used to estimate the sample expression. Differential expression gene (DEG) sequence was used for differential analysis, and Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) were used to annotate and analyze the functions of differential genes [40, 41]. Genes with q value < 0.5 and fold differences |FoldChange|> 2 were defined as significantly different genes.
Availability of data and materials
All the data generated in the study are included in the present manuscript. In addition, the reaction equations and DEGs have been listed in Additional files.
References
Najafpour G. Biochemical engineering and biotechnology. Amsterdam: Elsevier; 2006. p. 1–13.
Roca C, Nielsen J, Olsson L. Metabolic engineering of ammonium assimilation in xylose-fermenting Saccharomyes cerevisiae improves ethanol production. Appl Environ Microbiol. 2003;69:4732–6.
Hahn-Hagerdal B, Wahlbom CF, Gardonyi M, van Zyl WH, Cordero Otero RR, Jonsson LJ. Metabolic engineering of Saccharomyces cerevisiae for xylose utilization. Adv Biochem Eng Biotechnol. 2001;73:53–84.
Pronk JT, Yde Steensma H, Van Dijken JP. Pyruvate metabolism in Saccharomyces cerevisiae. Yeast. 1996;12:1607–33.
Morita K, Matsuda F, Okamoto K, Ishii J, Kondo A, Shimizu H. Repression of mitochondrial metabolism for cytosolic pyruvate-derived chemical production in Saccharomyces cerevisiae. Microb Cell Fact. 2019;18:1–7.
Fukuhara H. The Kluyver effect revisited. FEMS Yeast Res. 2003;3:327–31.
Nissen TL, Kielland-Brandt MC, Nielsen J, Villadsen J. Optimization of ethanol production in Saccharomyces cerevisiae by metabolic engineering of the ammonium assimilation. Metab Eng. 2000;2:69–77.
Bideaux C, Alfenore S, Cameleyre X, Molina-Jouve C, Uribelarrea JL, Guillouet SE. Minimization of glycerol production during the high-performance fed-batch ethanolic fermentation process in Saccharomyces cerevisiae, using a metabolic model as a prediction tool. Appl Environ Microbiol. 2006;72:2134–40.
Albertyn J, Hohmann S, Thevelein JM, Prior BA. GPD1, which encodes glycerol-3-phosphate dehydrogenase, is essential for growth under osmotic stress in Saccharomyces cerevisiae, and its expression is regulated by the high-osmolarity glycerol response pathway. Mol Cell Biol. 1994;14:4135–44.
Eriksson P, Andre L, Ansell R, Blomberg A, Adler L. Cloning and characterization of GPD2, a second gene encoding sn-glycerol 3-phosphate dehydrogenase (NAD+) in Saccharomyces cerevisiae, and its comparison with GPD1. Mol Microbiol. 1995;17:95–107.
Chen XL, Li SB, Liu LM. Engineering redox balance through cofactor systerms. Trends Biotechnol. 2014;32:337–43.
Heux S, Cachon R, Dequin S. Cofactor engineering in Saccharomyces cerevisiae: expression of a H2O-forming NADH oxidase and impact on redox metabolism. Metab Eng. 2006;8:303–14.
Russell DW, Smith M, Williamson VM, Young ET. Nucleotide sequence of the yeast alcohol dehydrogenase II gene. J Biol Chem. 1983;258:2674–82.
Ida Y, Furusawa C, Hirasawa T, Shimizu H. Stable disruption of ethanol production by deletion of the genes encoding alcohol dehydrogenase isozymes in Saccharomyces cerevisiae. J Biosci Bioeng. 2012;113:192–5.
Bastian S, Liu X, Meyerowitz JT, Snow CD, Chen MMY, Arnold FH. Engineered ketol-acid reductoisomerase and alcohol dehydrogenase enable anaerobic 2-methylpropan-1-ol production at theoretical yield in Escherichia coli. Metab Eng. 2011;13:345–52.
Hou J, Lages NF, Oldiges M, Vemuri GN. Metabolic impact of redox cofactor perturbations in Saccharomyces cerevisiae. Metab Eng. 2009;11:253–61.
Remize F, Barnavon L, Dequin S. Glycerol export and glycerol-3-phosphate dehydrogenase, but not glycerol phosphatase, are rate limiting for glycerol production in Saccharomyces cerevisiae. Metab Eng. 2001;3:301–12.
Tamas MJ, Luyten K, Sutherland FC, Hernandez A, Albertyn J, Valadi H, Li H, Prior BA, Kilian SG, Ramos J, et al. Fps1p controls the accumulation and release of the compatible solute glycerol in yeast osmoregulation. Mol Microbiol. 1999;31:1087–104.
Walz T, Hirai T, Murata K, Heymann JB, Mitsuoka K, Fujiyoshi Y, Smith BL, Agre P, Engel A. The three-dimensional structure of aquaporin-1. Nature. 1997;387:624–7.
Zhang A, Kong Q, Cao L, Chen X. Effect of FPS1 deletion on the fermentation properties of Saccharomyces cerevisiae. Lett Appl Microbiol. 2007;44:212–7.
Lee JY, Ishida Y, Takahashi T, Naganuma A, Hwang GW. Transport of pyruvate into mitochondria is involved in methylmercury toxicity. Sci Rep. 2016;6:1–9.
Nevoigt E. Progress in metabolic engineering of Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2008;72:379–412.
Nissen TL, Hamann CW, Kielland-Brandt MC, Nielsen J, Villadsen J. Anaerobic and aerobic batch cultivations of Saccharomyces cerevisiae mutants impaired in glycerol synthesis. Yeas. 2000;16:463–74.
Atsumi S, Hanai T, Liao JC. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature. 2008;451:86–13.
Cao LM, Zhang AL, Kong QX, Xu XJ, Josine TL, Chen X. Overexpression of GLT1 in fps1 Delta gpd Delta mutant for optimum ethanol formation by Saccharomyces cerevisiae. Biomol Eng. 2007;24:638–42.
Guo ZP, Zhang L, Ding ZY, Wang ZX, Shi GY. Interruption of glycerol pathway in industrial alcoholic yeasts to improve the ethanol production. Appl Microbiol Biotechnol. 2009;82:287–92.
Wisselink HW, Cipollina C, Oud B, Crimi B, Heijnen JJ, Pronk JT, van Maris AJA. Metabolome, transcriptome and metabolic flux analysis of arabinose fermentation by engineered Saccharomyces cerevisiae. Metab Eng. 2010;12:537–51.
Zhang GC, Kong II, Kim H, Liu JJ, Cate JHD, Jin YS. Construction of a quadruple auxotrophic mutant of an industrial polyploid Saccharomyces cerevisiae strain by using RNA-guided Cas9 nuclease. Appl Environ Microbiol. 2014;80:7694–701.
Monosik R, Magdolen P, Stredansky M, Sturdik E. Monitoring of monosaccharides, oligosaccharides, ethanol and glycerol during wort fermentation by biosensors, HPLC and spectrophotometry. Food Chem. 2013;138:220–6.
Bressani APP, Martinez SJ, Sarmento ABI, Borem FM, Schwan RF. Organic acids produced during fermentation and sensory perception in specialty coffee using yeast starter culture. Food Res Int. 2020;128:1–9.
Nissen TL, Schulze U, Nielsen J, Villadsen J. Flux distributions in anaerobic, glucose-limited continuous cultures of Saccharomyces cerevisiae. Microbiology. 1997;143:203–18.
Li H, Su J, Ma W, Guo AQ, Shan ZH, Wang H. Metabolic flux analysis of Saccharomyces cerevisiae in a sealed winemaking fermentation system. FEMS Yeast Res. 2015;15:1–8.
Morales Y, Bosque G, Vehi J, Pico J, Llaneras F. PFA toolbox: a MATLAB tool for metabolic flux analysis. BMC Syst Biol. 2016;10:1–7.
Chen YH, Zhang X, Zhang M, Zhu JY, Wu ZF, Zheng XJ. A transcriptome analysis of the ameliorate effect of Cyclocarya paliurus triterpenoids on ethanol stress in Saccharomyces cerevisiae. World J Microbiol Biotechnol. 2018;34:1–10.
Holm JB, Humphrys MS, Robinson CK, Settles ML, Ott S, Fu L, Yang HQ, Gajer P, He X, McComb E, et al. Ultrahigh-throughput multiplexing and sequencing of >500-base-pair amplicon regions on the Illumina HiSeq 2500 platform. Msystems. 2019;4:1–8.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Zhao Q, Pan LQ, Ren Q, Hu DX. Digital gene expression analysis in hemocytes of the white shrimp Litopenaeus vannamei in response to low salinity stress. Fish Shellfish Immunol. 2015;42:400–7.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15.
Wang LG, Wang SQ, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–5.
Wang LK, Feng ZX, Wang X, Wang XW, Zhang XG. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–8.
Du JL, Li ML, Yuan ZF, Guo MC, Song JZ, Xie XZ, Chen YL. A decision analysis model for KEGG pathway analysis. BMC Bioinform. 2016;17:1–8.
Acknowledgements
We acknowledge the technical assistance of Hongpu Wang from Hefei Agricultural and Rural Bureau for the determination of glucose and glycerol contents using the HPLC method. We also appreciate Kanglin Wang from Hefei Kangnuo Biopharmaceutical Co., Ltd for help in the molecular technology.
Funding
This study was funded by the Major Science and Technology Projects of Anhui Province (202003c08020001; 202103b06020014), Natural Science Foundation of Anhui Province (1908085MC80), and Natural Science Foundation of Hefei City (2022047).
Author information
Authors and Affiliations
Contributions
PZY provided conceptualization and performed writing; SYJ and SHL performed the experiment; SWJ provided software; STJ, YHD, JLL, and HW provided resources; YZ performed investigation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Reaction equations of metabolites in S. cerevisiae.
Additional file 2.
S. cerevisiae DEGs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, P., Jiang, S., Lu, S. et al. Ethanol yield improvement in Saccharomyces cerevisiae GPD2 Delta FPS1 Delta ADH2 Delta DLD3 Delta mutant and molecular mechanism exploration based on the metabolic flux and transcriptomics approaches. Microb Cell Fact 21, 160 (2022). https://doi.org/10.1186/s12934-022-01885-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-022-01885-3