
Abstract
Background
The gut microbiota is a complex ecosystem, which is essential for the metabolism, health and immunity of host. Many diseases have been shown to be closely related to the alteration of intestinal flora. Aeromonas veronii as a conditioned pathogen can cause disease in Yangtze finless porpoise through intestinal infections. However, it is not clear whether the disease caused by Aeromonas veronii is related to changes of intestinal flora. In the current study, the diversity and composition of gut microbiota in the healthy and Aeromonas veronii-infected Yangtze finless porpoise were evaluated by high-throughput sequencing to further investigate the potential association between intestinal flora alteration and pathogen invasion.
Results
A total of 127,3276 high-quality sequences were achieved and 2465 operational taxonomic units (OTUs) were in common among all samples. The results of alpha diversity showed that there was no obvious difference in richness and diversity between healthy and Aeromonas veronii-infected Yangtze finless porpoise. Firmicutes, Bacteroidetes and Proteobacteria were the most dominant phyla in all samples. In addition, the healthy Yangtze finless porpoise exhibited higher abundance of Firmicutes and Fusobacteria than Aeromonas veronii-infected Yangtze finless porpoise, while, the level of Proteobacteria was decreased. At the genus level, Paeniclostridium and Paraclostridium were the predominant bacteria genera in the CK (healthy Yangtze finless porpoise) group. In the DIS (Aeromonas veronii-infected Yangtze finless porpoise) group, Lactobacillus and unidentified_Enterobacteriaceae were the dominant bacteria genera and the proportion of Paeniclostridium, Paraclostridium, Terrisporobacter, Cetobacterium, Candidatus Arthromitus, Terrabacter and Dechloromonas were reduced.
Conclusions
In conclusion, our results showed that Aeromonas veronii infection can alter the gut microbiota of the Yangtze finless porpoise by affecting the number of harmful bacteria and beneficial bacteria.
Similar content being viewed by others

Introduction
The Yangtze finless porpoise (Neophocaena phocaenoides asiaeorientalis) is a rare species that mainly lives in the Yangtze River basin, Dongting lake and Poyang lake in China. Furthermore, the Yangtze finless porpoise is the only freshwater population of porpoises in the world. However, the survival of Yangtze finless porpoise has suffered serious threats due to decline in water quality and overfishing over the last several decades. According to statistics, the number of Yangtze finless porpoise is gradually declining and less than 2000 are remaining [1, 2]. The Yangtze finless porpoise has been listed as an endangered species by the International Union for Conservation of Nature (IUCN) since 2013. Multiple protection measures including captive breeding, in situ and ex situ conservation have been applied to prevent the continuous reduction of this unique porpoise since the end of the last century. At present, two semi-natural and seven natural reserves have been built.
Aeromonas spp., as one of the main pathogens of aquatic animals poses a huge threat to the health of aquatic animals [3, 4]. Liu et al. reported that Aeromonas veronii can cause the skin necrosis, visceral hemorrhage and even death of Yangtze finless porpoise [5]. Not only that, Aeromonas veronii can cause a variety of diseases in terrestrial animals and humans, such as dysentery, sepsis and necrotizing fasciitis [6]. Most of the studies suggest that Aeromonas veronii is an opportunistic pathogen that regulates the expression of virulence factors according to the surrounding environment [7, 8]. The latest research shows that the infection of Aeromonas veronii may interact with other bacteria and its pathogenicity may be related to the intestinal flora [9].
The intestinal bacterial community consists of a vast number of different microorganisms including commensals, pathogens and some conditioned pathogens [10, 11]. The intestinal flora plays a vital role in growth, metabolism and immunity of the host [12,13,14]. In addition, intestinal microorganisms can inhibit the proliferation of pathogenic bacteria in the host by competing with the pathogens for nutrients and adhesion sites [15, 16]. Meanwhile, some gut microbes can also produce several metabolites with bacteriostatic effects to prevent the reproduction of pathogenic bacteria [17]. The stability of intestinal flora is a prerequisite for the intestine to play a mechanical and immune barrier against the invasion of pathogenic microorganisms. However, the stability of intestinal flora can be affected by many intrinsic and extrinsic factors, including temperature, environment, antibiotic, and host phenotypes [18]. Previous studies have shown that intestinal flora alteration is closely related to many diseases including diarrhea, rheumatoid arthritis, diabetes and obesity [19,20,21]. More importantly, intestinal flora imbalance can lead to some conditional pathogens which may show strong pathogenicity [22, 23]. Previous studies have reported differences in intestinal microbes between healthy and Aeromonas veronii-infected grass carp [24]. However, still less is known about the intestinal flora structure in healthy and Aeromonas veronii-infected Yangtze finless porpoise. Therefore, the objective of the current study was to analyze the microbial diversity of healthy and Aeromonas veronii-infected Yangtze finless porpoise by high-throughput sequencing.
Materials and methods
Animals and sample collection
Yangtze finless porpoises used in the present study are raised in Anqing Nature Reserve (Anqing, China). The experimental animals (approximately 3 years old, and four males and four females in each group) were selected in the same water area (Table 1). The clinical symptoms of the diseased Yangtze finless porpoise were depression, conjunctival hemorrhage and skin necrosis. Clinical observation, histopathological examination, bacterial isolation and identification, PCR amplification and gene sequence alignment were used to evaluate etiological agent and ultimately determined that the disease in Yangtze finless porpoises was due to Aeromonas veronii infection. The relevant research about disease assessment has been published in the Diseases of Aquatic Organisms [5]. Moreover, the Yangtze finless porpoises in the control group were declared healthy after being examined by the professional veterinarian.
The medical infusion tube (cut the both ends of the medical infusion tube by scissor) dipped in a small amount of petroleum jelly was slowly and rotationally inserted into the anus 10–15 cm of the Yangtze finless porpoise to obtain fecal samples. In this study, a total of 16 fresh fecal samples were collected from sixteen Yangtze finless porpoise (healthy Yangtze finless porpoise: CK1, CK2, CK3, CK4, CK5,CK6, CK7, CK8 and Aeromonas veronii-infected Yangtze finless porpoise: DIS1, DIS2, DIS3, DIS4, DIS5, DIS6, DIS6, DIS7, DIS8) by using sterile tool. All the samples were stored in sterile plastic and transported to the laboratory in ice boxes and then stored at − 80 °C for further study.
gDNA extraction
Prior to the gDNA extraction, all the samples need to be preprocessed. Initially, the intestinal contents were washed three times with phosphate buffer solution and then the PBS-washed intestinal contents were centrifuged at 500 rpm for 4 min to obtain the sediments. Afterwards, the obtained sediments were resuspended with PBS to further study. The gDNA of each sample was extracted via QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany) according to manufacturer’s recommendations. The extraction quality of gDNA was evaluated by using 0.8% agarose gel electrophoresis. Simultaneously, the Nanodrop™spectrophotometer (NanoDrop Technologies, Thermo Scientific, USA) was used to quantify the concentration of gDNA.
16S rRNA gene amplification and sequencing
Universal primers (338F: ACTCCTACGGGAGGCAGCA and 806R: GGACTACHVGGGTWTCTAAT) with barcode were synthesized based on conserved regions in the sequence to amplify the V3–V4 regions of the 16S rRNA. The 2% agarose gel electrophoresis and gel recovery kit (AXYGEN, USA) were used for PCR amplification product evaluation and target segment recovery, respectively. The PCR-recycled products were quantified on Microplate reader (BioTek, FLx800) using Quant-iT PicoGreen dsDNA Assay Kit. The TruSeq Nano DNA LT Library Prep Kit (Illumina, USA) was used to prepare the sequencing library. The sequence ends of the above-mentioned amplified products were repaired by using End Repair Mix2. The self-connected fragments of linker were removed via using magnetic bead screening and the sequencing library with linker was purified. The above DNA fragments with linker were PCR amplified to enrich the sequencing library and BECKMAN AMPure XP Beads was used to purify the library enrichment product. The same volume of 1 × loading buffer (contained SYBR green) was mixed with the PCR products, and detection was performed by using 2% agarose gel electrophoresis on electrophoresis system (DYCZ-20A, Beijing, China). Moreover, The PCR products were mixed in equidensity ratios and purified using GeneJETTM Gel Extraction Kit (Thermo scientific).
Prior to the sequencing, the sequencing libraries were required to be inspected on Agilent Bioanalyzer via using Agilent High Sensitivity DNA Kit. The qualified library has only one peak and no linker. Furthermore, the Quant-iT PicoGreen dsDNA Assay Kit was used to quantify the libraries by the PromegaQuantiFluor fluorescence quantification system and the library concentrations above 2 nM were considered qualified. The qualified libraries were subjected to high-throughput sequencing after gradient dilution and NaOH denaturation into single strands. The raw sequence data has been submitted to the NCBI Sequence Read Archive (SRA) under accession no. PRJNA623474.
Bioinformatics and statistical analysis
The initial data of high-throughput sequencing was subjected to screening based on the sequence quality via using QIIME software. The sequence was identified and allocated into the corresponding sample based on the primers and barcode information and removed the chimera and interrogative sequences. The obtained sequences with 97% similarity were merged to the same operational taxonomic units (OTUs), and the representative sequences were used for phylogenetic analysis and taxonomic status identification. The representative sequence of each OTU was taxonomically classified based on the Ribosomal Database Project (RDP) database. The diversity of each sample was evaluated based on the abundance distribution of OTU in different samples and the sparse curves were used to assess the depth of sequencing. Four indexes including ACE, Shannon, Simpson and Chao1 were calculated to assess the alpha diversity. In addition, beta diversity analyses (Principal coordinate analysis) was performed to evaluate the similarity of community structure between different samples. GraphPad Prism 7 and R (v3.0.3) software were used for data analysis. Additionally, p-values less than 0.05 were considered statistically significant and the values were presented as mean ± SD.
Results
Sequences analyses
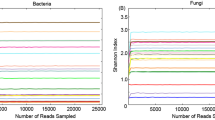
It is well known that a number of erroneous or questionable sequences can be produced during the high throughput sequencing. Therefore, the effective sequences were further evaluated and filtered to obtain reliable sequences which can be used for subsequent analysis. The length of qualified sequence should be greater than 150 bp and ambiguous base N was also not allowed. Moreover, the sequences with > 1 mismatched bases at 5′ end or contained > 8 same bases in succession were discarded. In the present study, a total of 127,3276 high-quality sequences were obtained from 16 samples. Additionally, the number of valid sequences from CK group ranged from 69,077 to 85,150, while the number of valid sequences from DIS group ranged from 80,055 to 80,270 (Table 2). The Chao1 and Shannon curves in each sample were extended all the way to the right end of the x-axis and the Rank abundance curve exhibited high and long slight degree broken line. The results of rarefaction curves (Chao1 curve and Shannon curve) and rank abundance curve suggested that the total number of sequences, depth, abundance and evenness meet the requirement for sequencing and analysis (Figs. 1, 2). The OTUs were recognized on the basis of 97% nucleotide-sequence similarity. Therefore, a total of 3806 and 3246 OTUs were identified in the CK and DIS groups, respectively, and 2465 OTUs in common (Fig. 3a). Meanwhile, 160 and 136 core OTUs were found in the CK and DIS groups, respectively (Fig. 3b, c). Additional file 1: Table S1 showing the number of OTUs identified for each sample at different levels.

Feasibility analysis of different samples. Each curve indicates a sample. The rarefaction curves (A, B) were used to assess the adequacy of sequencing for each sample. Rank abundance curve (C) was used to evaluate the evenness and abundance of samples. CK indicates the healthy Yangtze finless porpoise, while DIS represents the Yangtze finless porpoise infected by Aeromonas veronii

The rank abundance curve of different samples. The rank abundance curve was used to evaluate the evenness and abundance of samples

Venn diagrams of the OTUs composition. a Venn diagrams of comparison in CK and DIS groups. b Venn diagrams for core OTUs composition in the CK group. c Venn diagrams for core OTUs composition in the DIS group. CK indicates the healthy Yangtze finless porpoise, while DIS represents the Yangtze finless porpoise infected by Aeromonas veronii
Analysis of microbial community diversity in different groups
In the current study, Chao1, ACE, Shannon and Simpson were used to evaluate the alpha diversity of the intestinal microbial community (Table 3). The average of Chao1 and ACE indices in DIS group (1362.56 and 1376.007) were higher than those of CK group (1272.937 and 1278.566), while no significant difference was observed between the two groups (P > 0.05) (Fig. 4a, b). The results of Chao 1 and ACE indices suggested that there was no significant difference in the intestinal flora abundance between the two groups. The average of Shannon index was 5.861 and 6.41 in CK group and DIS group, respectively, with no significant difference between two groups (P > 0.05) (Fig. 4c). The average of Simpson index was 0.937 in CK group, 0.929 in DIS group, with no obvious difference in the two groups (P > 0.05) (Fig. 4d). The Shannon and Simpson indices revealed that the difference in the flora evenness between CK and DIS group was non-significant. The PCoA analysis clearly showed the difference among all sample individuals or groups. The results demonstrated that all the samples clustered closely into the two categories, whereas the DIS1.4 and DIS1.8 were specific (Fig. 5).

The diversity indices of intestinal microbial community in different groups. The alpha diversity of intestinal flora can be reflected by the Chao1 (a), ACE (b), Shannon (c) and Simpson (d). CK and DIS indicate the healthy and Aeromonas veronii-infected Yangtze finless porpoise, respectively

Principal coordinate analysis (PCoA) of intestinal flora. Each dot indicates one sample. The distance of the two points indicates the difference of gut microbiota. CK and DIS refer to healthy and Aeromonas-infected Yangtze finless porpoise, respectively
Composition analysis of the microbial community structure in different groups
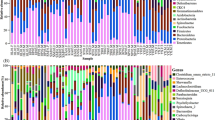
The intestinal microbial community structure in disease and healthy Yangtze finless porpoises were evaluated at different levels, respectively. In the CK group, the preponderant bacteria at phylum level were Firmicutes (71.9%), Proteobacteria (7.4%), Bacteroidetes (8.2%) and Tenericutes (4.7%) and the sum of abundance was over 92% (Fig. 6a). Other phyla including Oxyphotobacteria (1.2%), Acidobacteria (0.4%), Chloroflexi (0.2%) and Spirochaetes (0.4%) were represented with a lower abundance and the total abundance was less than 3% (Fig. 6a). In the DIS group, Firmicutes (38.1%), Proteobacteria (27.0%), Bacteroidetes (20.0%) and Actinobacteria (5.7%) were the four most dominant phyla with a little difference from the control group (Fig. 6a). At the level of genus, the top 30 dominant genera in all collected samples are shown in Fig. 6b. Specifically, Paeniclostridium (23.7%), Romboutsia (10.4%), Lactobacillus (3.9%) and Paraclostridium (13.1%) were the predominant bacteria genera in the CK group. Meanwhile, Lactobacillus (11.0%), unidentified_Clostridiales (7.4%), unidentified_Enterobacteriaceae (8.3%) and Romboutsia (5.5%) were observed as the predominant in the DIS group. Remarkably, unidentified_Clostridiales (33.7% and 20.0%) and unidentified_Enterobacteriaceae (30.1% and 34.4%) were the most predominant genera in the DIS1.4 and DIS1.8, respectively.

The relative abundance of microbial composition of different samples. a The top 10 dominant phylum of the Yangtze finless porpoise intestinal flora. b The top 30 primary genera of the Yangtze finless porpoise intestinal flora. CK and DIS refer to healthy and Aeromonas veronii-infected Yangtze finless porpoise, respectively
The comparison of intestinal bacterial communities between CK and DIS groups were also performed at the levels of phylum and genus. The results suggested that at the phylum level the abundance of Firmicutes (P < 0.001) and Fusobacteria (P < 0.001) in the CK groups were significantly higher than the DIS group, while the Proteobacteria (P < 0.05) content was lower (Fig. 7a). As for genus level, Paeniclostridium (P < 0.001), Paraclostridium (P < 0.001), Terrisporobacter (P < 0.001), Cetobacterium (P < 0.001), Candidatus Arthromitus (P < 0.05), Terrabacter (P < 0.05) and Dechloromonas (P < 0.05) were more preponderant in the CK group than the DIS group (Fig. 7b, c).

Differences in intestinal bacteria abundance between the CK and DIS groups. a Differences in phylum abundance between the CK and DIS groups. b, c Differences in genus abundance between the CK and DIS groups. CK: the healthy Yangtze finless porpoise. DIS: the Yangtze finless porpoise infected by Aeromonas veronii. The results were evaluated through one-way ANOVA. All of the data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
It is well known that the gut microbial community is an interactive and complex system which has a great influence on the host. Intestinal microbial community is an important barrier for the organism to resist the invasion and colonization of foreign pathogens that plays a key role in the prevention and treatment of diseases. Therefore, it is meaningful to conduct studies on the composition of intestinal flora in different species. Up till now, a large amount of studies has been performed to investigate the role of intestinal flora in a wide range of dysfunctions and diseases [25, 26]. With rapid advancement and development of high-throughput sequencing technology, the composition of gut microbiota community has been investigated in many species, including chicken, camel, piglet, grass carp and cattle [27,28,29]. However, few studies have focused on the relationship between microbial community structure and disease of Yangtze finless porpoise induced by Aeromonas veronii [5]. In the current study, we made a comparison of intestinal flora communities between healthy and Aeromonas veronii-infected Yangtze finless porpoise by using high-throughput sequencing.
Considering the scarcity of the species, we selected fecal samples as the research object to evaluate the diversity of intestinal microorganisms. Our results suggested that the number of OTU in the healthy Yangtze finless porpoise was higher than that in the Aeromonas veronii-infected Yangtze finless porpoise. However, the results of Chao1, ACE, Shannon and Simpson showed that there was no obvious difference in the abundance and evenness of microbial diversity in Yangtze finless porpoise of different groups, which was consistent with previous studies in the zebrafish with intestinal flora imbalance [30].
The interaction of microbe in the intestines can lead to a huge influence on the immunity, nutrition and health of the organism [31]. It is well-known that Firmicutes, Proteobacteria and Bacteroidetes are the most dominant phyla of the mammals and the percentage of each phylum can be affected by animals’ species, environment, feed [32]. In the current study, the dominant phyla were Firmicutes, Proteobacteria and Bacteroidetes in all the samples, which was in line with many studies in intestinal flora of mammals [33,34,35]. However, there were significant differences in Firmicutes and Proteobacteria between the CK and DIS groups. Proteobacteria is the largest phylum, which contains a large number of gram-negative pathogenic bacteria, such as Vibrio cholerae, Salmonella spp., Helicobacter Pylori and Escherichia coli [36, 37]. The higher abundance of Proteobacteria in the intestines increase the risk of pathogen infection. Firmicutes are mainly composed of some gram-positive bacteria including Lactobacillus spp., Listeria spp. and Lactococcus spp. [38]. Some studies have shown that Firmicutes were closely related to carbohydrate and protein digestion [39,40,41]. The high abundance of Firmicutes in the intestine will contribute to meet the nutritional and energy requirements of host during growth and development [39, 40]. Lactobacillus spp., Listeria spp. and Lactococcus spp. are considered probiotics and play an important role in resisting pathogenic bacteria, maintaining intestinal flora balance and improving immunity [42, 43]. Compared with the CK group, the DIS group had lower Firmicutes content and higher Proteobacteria level, which indicated that the intestinal flora of the Aeromonas veronii-infected Yangtze finless porpoise was imbalanced. Generally, the composition of gut microbiota will change with the influence of external environment to some extent [44]. Although, the intestinal microbial community is in dynamic changes, it may still maintain its functional stability due to the existence of a large number of functionally redundant species [45]. However, significant changes in microbial community will cause intestinal flora imbalance and affect its function. Previous studies have suggested that the observable changes in intestinal flora of young aquatic animals may be one of the reasons for the high mortality [46]. Furthermore, fish with intestinal flora alternation are more susceptible to invasion by pathogenic microorganisms [47, 48].
Cetobacterium spp. mostly inhabits in the intestines of fish and contribute to fermentation of peptides and carbohydrate [49, 50]. Additionally, Cetobacterium spp. has already been reported to produce vitamin B12 and was considered an important symbiotic organism that provides vitamin B12 to the host [51, 52]. Vitamin B12 can only be synthesized by microorganisms in the host. Vitamin B12 can promote the development and maturation of red blood cells, prevent pernicious anemia and maintain the health of the nervous system. Prior research has shown that the abundance of genus Cetobacterium was significantly decreased in graphene-induced intestinal microflora imbalance in zebrafish and cadmium-induced intestinal flora alteration in carassius auratus gibelio [53, 54]. Ma et al. indicated that the level of Cetobacterium was significantly decreased in Yunlong Grouper with intestinal microbiota alteration [55]. Moreover, Parshukov et al. showed that the relative abundance of Cetobacterium in bacteria infected Oncorhynchus mykiss was significantly lower than in healthy Oncorhynchus mykiss [56]. In the present study, the abundance of Cetobacterium in the DIS group was significantly reduced, which is in accordance with the previous studies of intestinal flora imbalance [53, 54]. Candidatus Arthromitus spp., as a gut symbiotic bacterium, plays a vital role in the maturation of the host’s immune system [57]. The lower abundance of genus Candidatus Arthromitus in the DIS group, which suggested that the immune system in the Aeromonas veronii-infected Yangtze finless porpoise may be affected. In addition, we also found some bacteria related to degradation of pollutants in the CK group. Terrabacter spp. is well-known for its versatility in degrading many typical persistent organic pollutants [58, 59]. Dechloromonas spp. has reducing capacity and can reduce metal contaminants in water [60, 61]. Therefore, Dechloromonas spp. can be used to alleviate water pollution caused by a variety of metal compounds. We speculate that genera Terrabacter and Dechloromonas in the intestines may contribute to mitigation the toxic effects of pollutants in water. Remarkably, the relative abundance of genera Enterobacter and Clostridium in the DIS1.4 and DIS1.8 were higher than those in the other samples. Clostridium spp. was closely related to intestinal toxemia and diarrhea in mammals and its toxins affect the host health by different pathways [62]. Additionally, Clostridium spp. has also been shown to play an important role in causing necrotizing enterocolitis in preterm infants [63]. It has been reported that Enterobacter spp. contributed to the occurrence of bacteremia [64]. Therefore, the higher abundance of genera Enterobacter and Clostridium in the gut may increase the risk of intestinal toxemia, diarrhea and bacteremia.
In conclusion, the current study has compared the differences of diversity and composition about gut flora structure between healthy and Aeromonas veronii-infected Yangtze finless porpoise for the first time. The results suggested that there was no significant difference in abundance of intestinal flora between the two groups. The abundance of Firmicutes and Fusobacteria in the Yangtze finless porpoise were significantly decreased, while, the abundance of Proteobacteria was significantly increased after Aeromonas veronii infection. Furthermore, Aeromonas veronii altered the primary intestinal flora composition in Yangtze finless porpoise by decreasing the relative abundance of genera Paeniclostridium, Paraclostridium, Terrisporobacter, Cetobacterium and Candidatus Arthromitus. Importantly, we also observed that the levels of genera Terrabacter and Dechloromonas were reduced in the Aeromonas veronii-infected Yangtze finless porpoise, which can alleviate the toxic effects of pollutants in water. However, the gut microbiota can be affected by external environment (water quality and temperature), host diet, host requirement and age. Therefore, we cannot eliminate all the influential elements due to sample size and experiment conditions. However, our study indicated that Aeromonas veronii infection caused alterations in the gut microbiota of the Yangtze finless porpoise. This provides a new consideration for the prevention and treatment of Yangtze finless porpoise disease.
Availability of data and materials
Yes.
References
Zhao X, Barlow J, Taylor B, Pitman R, Wang K, Wei Z, Stewart B, Turvey S, Akamatsu T, Reeves R, Wang D. Abundance and conservation status of the Yangtze finless porpoise in the Yangtze River, China. Biol Conserv. 2008;141(12):3018.
Zhang X, Wang K. Population viability analysis for the Yangtze finless porpoise. Acta Ecol Sin. 1999;19(4):529–33.
Janda J, Abbott S. The Genus Aeromonas: taxonomy, pathogenicity, and infection. Clin Microbiol Rev. 2010;23(1):35–73.
Zhang X, Yang W, Wu H, Gong X, Li A. Multilocus sequence typing revealed a clonal lineage of Aeromonas hydrophila caused motile Aeromonas septicemia outbreaks in pond-cultured cyprinid fish in an epidemic area in central China. Aquaculture. 2014;432:1–6.
Liu Z, Zheng A, Chen M, Lian Y, Zhang X, Zhang S, Yu D, Li JK. Isolation and identification of pathogenic Aeromonas veronii from a dead Yangtze finless porpoise. Dis Aquat Org. 2018;132(1):13–22.
Monette S, Dallaire A, Mingelbier M, Groman D, Uhland C, Richard J, Paillard G, Johannson L, Chivers D, Ferguson HW, Leighton F, Simko E. Massive mortality of common carp (Cyprinus carpio carpio) in the St. Lawrence river in 2001: diagnostic investigation and experimental induction of Lymphocytic Encephalitis. Vet Pathol. 2006;43(3):302–10.
Merino S, Camprubí S, Tomás J. Effect of growth temperature on outer membrane components and virulence of Aeromonas hydrophila strains of serotype O:34. Infect Immun. 1992;60(10):4343–9.
Gonzalez-Serrano C, Santos J, García-López M, Otero A. Virulence markers in Aeromonas hydrophila and Aeromonas veronii biovar sobria isolates from freshwater fish and from a diarrhoea case. J Appl Microbiol. 2002;93(3):414–9.
Thornley J, Shaw J, Gryllos I, Eley A. Virulence properties of clinically significant Aeromonas species: evidence for pathogenicity. Rev Med Microbiol. 1997;8(2):61–72.
Abudabos A, Alyemni A, Dafalla Y, Khan R. Effect of organic acid blend and bacillus subtilis alone or in combination on growth traits, blood biochemical and antioxidant status in broilers exposed to Salmonella typhimurium challenge during the starter phase. J Appl Anim Res. 2017;45:1–5.
Sidhu M, Van D. The gut microbiome. Aust Fam Phys. 2017;46:206.
Shapira M. Gut microbiotas and host evolution: scaling up symbiosis. Trends Ecol Evol. 2016;31:539–49.
Smith P. The tantalizing links between gut microbes and the brain. Nature. 2015;526:312–4.
Mangiola F, Ianiro G, Franceschi F, Fagiuoli S, Gasbarrini G, Gasbarrini A. Gut microbiota in autism and mood disorders. World J Gastroenterol. 2016;22:361–8.
Decamp O, Moriarty D, Lavens P. Probiotics for shrimp larviculture: review of field data from Asia and Latin America. Aquac Res. 2008;39(4):334–8.
Pham T, Lawley T. Emerging insights on intestinal dysbiosis during bacterial infections. Curr Opin Microbio. 2014;17:67–74.
Popova M, Molimard P, Courau S, Crociani J, Dufour C, Le V, Carton T. Beneficial effects of probiotics in upper respiratory tract infections and their mechanical actions to antagonize pathogens. J Appl Microbiol. 2012;113(6):1305–18.
Paola M, Filippo C, Cavalieri D, Ramazzotti M, Poullet J, Massart S, Collini S, Pieraccini G, Lionetti P. PP90 impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Digest Liver Dis. 2011;43:445–6.
Zhang P, Meng X, Li D, Calderone R, Mao D, Sui B. Commensal homeostasis of gut microbiota-host for the impact of obesity. Front Physiol. 2017;8:1122.
Li X, Watanabe K, Kimura I. Gut microbiota dysbiosis drives and implies novel therapeutic strategies for diabetes mellitus and related metabolic diseases. Front Immunol. 2017;8:1882.
Sartor R. Review article: role of the enteric microflora in the pathogenesis of intestinal inflammation and arthritis. Aliment Pharmacol Ther. 1997;11:17–22.
Clemente J, Ursell L, Parfery L, Knight R. The impact of the gutmicrobiota on human health: an integrative view. Cell. 2012;148:1258–70.
Huang Z, Li X, Wang L, Shao Z. Changes in the intestinal bacterial community during the growth of white shrimp, Litopenaeus vannamei. Aquac Res. 2016;47:1737–46.
Huang H, Zhou P, Chen P, Xia L, Hu S, Yi G, Lu J, Yang S, Xie J, Peng J, Ding X. Alteration of the gut microbiome and immune factors of grass carp infected with Aeromonas veronii and screening of an antagonistic bacterial strain (Streptomyces flavotricini). Microb Pathog. 2020;143:104092.
Sherman M, Zaghouani H, Niklas V. Gut microbiota, the immune system, and diet influence the neonatal gut-brain axis. Pediatr Res. 2015;77(1–2):127.
Tang W, Kitai T, Hazen S. Gut microbiota in cardiovascular health and disease. Circ Res. 2017;120(7):1183–96.
Meale S, Li S, Azevedo P, Derakhshani H, DeVries T, Plaizier J, Steele M, Khafipour E. Weaning age influences the severity of gastrointestinal microbiome shifts in dairy calves. Sci Rep. 2017;7(1):198.
Chen L, Xu Y, Chen X, Fang C, Zhao L, Chen F. The maturing development of gut microbiota in commercial piglets during the weaning transition. Front Microbiol. 2017;8:1688.
Chupani L, Barta J, Zuskova E. Effects of food-borne ZnO nanoparticles on intestinal microbiota of common carp (Cyprinus carpio L.). Environ Sci Pollut Res Int. 2019;26(25):25869–73.
Zheng M, Lu J, Lin G, Su H, Sun J, Luan T. Dysbiosis of gut microbiota by dietary exposure of three graphene-family materials in zebrafish (Danio rerio). Environ Pollut. 2019;254:112969.
Allesina S, Tang S. Stability criteria for complex ecosystems. Nature. 2011;483(7388):205–8.
Dibaise J, Zhang H, Crowell M, Krajmalnik-Brown R, Decker G, Rittmann B. Gut microbiota and its possible relationship with obesity. Mayo Clin Proc. 2008;83:460–9.
Jami E, Israel A, Kotser A, Mizrahi I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013;7:1069–79.
Han Z, Li K, Shahzad M, Zhang H, Luo H, Qiu G, Lan Y, Wang X, Mehmood K, Li J. Analysis of the intestinal microbial community in healthy and diarrheal perinatal yaks by high-throughput sequencing. Microb Pathog. 2017;111:60–70.
Li A, Wang Y, Pei L, Mehmood K, Li K, Qamar H, Iqbal M, Waqas M, Liu J, Li J. Influence of dietary supplementation with Bacillus velezensis on intestinal microbial diversity of mice. Microb Pathog. 2019;136:103671.
Yang H, Xiao Y, Gui G, Li J, Wang J, Li D. Microbial community and short-chain fatty acid profile in gastrointestinal tract of goose. Poult Sci. 2018;97(4):1420–8.
Huang Z, Kraus V. Does lipopolysaccharide-mediated inflammation have a role in OA? Nat Rev Rheumatol. 2016;12(2):123–9.
Garneau J, Tremblay D, Moineau S. Characterization of 1706, a virulent phage from Lactococcus lactis with similarities to prophages from other Firmicutes. Virology. 2008;373(2):298–309.
Cheryl S, Greg W, Jeffrey S. Characterization of the primary starch utilization operon in the obligate anaerobe Bacteroides fragilis: regulation by carbon source and oxygen. J Bacteriol. 2006;188:4663.
Sun B, Wang X, Bernstein S, Huffman M, Xia D, Gu Z, Chen R, Sheeran L, Wagner R, Li J. Marked variation between winter and spring gut microbiota in free-ranging Tibetan Macaques (Macaca thibetana). Sci Rep. 2016;6:26035.
Zhang L, Jiang X, Li A, Waqas M, Gao X, Li K, Xie G, Zhang J, Mehmood K, Zhao S, Wangdui B, Li J. Characterization of the microbial community structure in intestinal segments of yak (Bos grunniens). Anaerobe. 2020;6:1102115.
Castillo N, Perdigon G, De Moreno de Le Blanc A. Oral administration of a probiotic Lactobacillus modulates cytokine production and TLR expression improving the immune response against Salmonella enterica serovar typhimurium infection in mice. BMC Microbiol. 2011;11:177–89.
Shimazu T, Villena J, Tohno M, Fujie H, Hosoya S, Shimosato T, Aso H, Suda Y, Kawai Y, Saito T, Makino S, Ikegami S, Itoh H, Kitazawa H. Immunobiotic Lactobacillus jensenii elicits anti-inflammatory activity in porcine intestinal epithelial cells by modulating negative regulators of the Tolllike receptor signaling pathway. Infect Immun. 2012;80:276–88.
Curtis T, Sloan W. Prokaryotic diversity and its limits: microbial community structure in nature and implications for microbial ecology. Curr Opin Microbio. 2004;7(3):221–6.
Cabrol L, Malhautier L. Integrating microbial ecology in bioprocess understanding: the case of gas biofiltration. Appl Microbiol Biot. 2011;90(3):837–49.
Rawls J, Mahowald M, Ley R, Gordon J. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell. 2006;127:423–33.
Qi X, Tu X, Zha J, Huang A, Wang G, Ling F. Immunosuppression-induced alterations in fish gut microbiota may increase the susceptibility to pathogens. Fish Shellfish Immu. 2019;88:540–5.
Robinson C, Bohannan B, Young V. From structure to function: the ecology of host-associated microbial communities. Microbiol Mol Biol R. 2010;74:453–76.
Li T, Long M, Gatesoupe F, Zhang Q, Li A, Gong X. Comparative analysis of the intestinal bacterial communities in different species of carp by pyrosequencing. Microb Ecol. 2015;69(1):25–36.
Finegold S, Vaisanen M, Molitoris D, Tomzynski T, Song Y, Liu C, Collins M, Lawson P. Cetobacterium somerae sp. nov. from human feces and emended description of the genus Cetobacterium. Syst Appl Microbiol. 2003;26(2):177–81.
Larsen A, Mohammed H, Arias C. Characterization of the gut microbiota of three commercially valuable warmwater fish species. J Appl Microbiol. 2014;116:1396–404.
Tsuchiya C, Sakata T, Sugita H. Novel ecological niche of Cetobacterium somerae, an anaerobic bacterium in the intestinal tracts of freshwater fish. Lett Appl Microbiol. 2008;46:43–8.
Wang N, Jiang M, Zhang P, Shu H, Li Y, Guo Z, Li Y. Amelioration of Cd-induced bioaccumulation, oxidative stress and intestinal microbiota by Bacillus cereus in Carassius auratus gibelio. Chemosphere. 2019;245:125613.
Siddik M, Chaklader M, Foysal M, Howieson J, Fotedar R, Gupta S. Influence of fish protein hydrolysate produced from industrial residues on antioxidant activity, cytokine expression and gut microbial communities in juvenile barramundi Lates calcarifer. Fish Shellfish Immunol. 2019;97:465–73.
Ma C, Chen C, Jia L, He X, Zhang B. Comparison of the intestinal microbiota composition and function in healthy and diseased Yunlong Grouper. AMB Express. 2019;9(1):187.
Parshukov A, Kashinskaya E, Simonov E, Hlunov O, Izvekova G, Andree K, Solovyev M. Variations of the intestinal gut microbiota of farmed rainbow trout, Oncorhynchus mykiss (Walbaum), depending on the infection status of the fish. J Appl Microbiol. 2019;127(2):379–95.
Yamauchi K, Snel J. Transmission electron microscopic demonstration of phagocytosis and intracellular processing of segmented filamentous bacteria by intestinal epithelial cells of the chick ileum. Infect Immun. 2000;68(11):6496–504.
Tappe W, Hofmann D, Disko U, Koeppchen S, Kummer S, Vereecken H. A novel isolated Terrabacter-like bacterium can mineralize 2-aminopyrimidine, the principal metabolite of microbial sulfadiazine degradation. Biodegradation. 2015;26(2):139–50.
Habe H, Chung J, Ishida A, Kasuga K, Die K, Takemura T, Nojiri H, Yamane H, Omori T. The fluorine catabolic linear plasmid in Terrabacter sp strain DBF63 carries the β-ketoadipate pathway genes, pcaRHGBDCFIJ, also found in proteobacteria. Microbiology. 2005;151:3713–22.
Terashima M, Yama A, Sato M, Yumoto I, Kamagata Y, Kato S. Culture-dependent and -independent identification of polyphosphate-accumulating dechloromonas spp. predominating in a full-scale oxidation ditch wastewater treatment plant. Microbes Environ. 2016;31:ME16097.
Zhang Y, Frankenberger W. Supplementing Bacillus sp. RS1 with Dechloromonas sp. HZ for enhancing selenate reduction in agricultural drainage water. Sci Total Environ. 2007;372(2–3):397–405.
Lewis C, Naylor R. Sudden death in sheep associated with Clostridium sordellii. Vet Rec. 1998;142:417–21.
Zhou J, He Z, Yang Y, Deng Y, Tringe S, Alvarezcohen L. High-throughput metagenomic technologies for complex microbial community analysis: open and closed formats. MBio. 2015;6:e02288.
Chow J, Fine M, Shlaes D, Quinn J, Hooper D, Johnson M, Ramphal R, Wagener M, Miyashiro D, Yu V. Enterobacter bacteremia: clinical features and emergence of antibiotic resistance during therapy. Ann Intern Med. 1991;115(8):585–90.
Acknowledgements
This work was supported by the Anhui University Provincial Natural Science Research Project (No. KJ2017A357) and Key Research and Development Plan of Anhui Province (No. 201904e01020013).
Author information
Authors and Affiliations
Contributions
ZGL and AL conceived and designed the experiments; YW, AZ, MMZ, ZKL, NW, CW, DPY contributed sample collection and reagents preparation. ZGL wrote the manuscript; AL and MI revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Animal experiments were performed under the approval of Ethics Committee of the Huazhong Agricultural University (Permit No. 4200695757).
Consent for publication
Yes.
Competing interests
There is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, Z., Li, A., Wang, Y. et al. Comparative analysis of microbial community structure between healthy and Aeromonas veronii-infected Yangtze finless porpoise. Microb Cell Fact 19, 123 (2020). https://doi.org/10.1186/s12934-020-01383-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-020-01383-4