
Abstract
Background
Erythritol is a natural sweetener that is used in the food industry. It is produced as an osmoprotectant by bacteria and yeast. Due to its chemical properties, it does not change the insulin level in the blood, and therefore it can be safely used by diabetics. Previously, it has been shown that erythrose reductase (ER), which catalyzes the final step, plays a crucial role in erythritol synthesis. ER reduces erythrose to erythritol with NAD(P)H as a cofactor. Despite many studies on erythritol synthesis by Yarrowia lipolytica, the enzymes involved in this metabolic pathway have ever been described.
Results
The gene YALI0F18590g encoding the predicted erythrose reductase from Y. lipolytica was overexpressed, and its influence on erythritol synthesis was studied. The amino acid sequence of the Y. lipolytica ER showed a high degree of similarity to the previously described erythrose reductases from known erythritol producers, such as Candida magnoliae and Moniliella megachiliensis. Here, we found that the gene overexpression results in an enhanced titer of erythritol of 44.44 g/L (20% over the control), a yield of 0.44 g/g and productivity of 0.77 g/L/h. Moreover, on purification and characterization of the enzyme we found that it displays the highest activity at 37 °C and pH 3.0. The effects of various metal ions (Zn2+, Cu2+, Mn2+, Fe2+) on erythrose reductase were investigated. The addition of Zn2+ ions at 0.25 mM had a positive effect on the activity of erythrose reductase from Y. lipolytica, as well as on the erythritol production.
Conclusions
In this study we identified, overexpressed and characterized a native erythrose reductase in Y. lipolytica. Further optimizations of this strain via metabolic pathway engineering and media optimization strategies enabled 54 g/L to be produced in a shake-flask experiment. To date, this is the first reported study employing metabolic engineering of the native gene involved in the erythritol pathway to result in a high titer of the polyol. Moreover, it indicates the importance of environmental conditions for genetic targets in metabolic engineering.
Similar content being viewed by others

Background
Erythritol is a natural four-carbon polyol which occurs in wine, honey and fermented food. Due to its sweetness (70% of the sweetness of sucrose), erythritol has been used as a low-calorie sweetener for decades [1]. In humans, erythritol is not metabolized, but excreted by renal processes. Consequently, it has very limited potential to induce changes in insulin levels, and therefore it can be consumed by diabetics [2]. Its maximum no-effect dose for causing diarrhea is the highest among polyols, that is why it does not cause gastrointestinal side effects [3]. Erythritol can be synthesized from dialdehyde starch by a high-temperature chemical reaction in the presence of a metal catalyst, but this chemical process involves several steps, and therefore production costs are high [4]. For these reasons, on the industrial scale erythritol has been produced from glucose in biotechnological processes by osmophilic yeast such as Trichosporonoides sp. [5], Candida magnoliae [6] or Moniliella sp. [7]. It was shown that erythritol is also produced by the yeast Yarrowia lipolytica [8]. Y. lipolytica has been granted “generally recognized as safe” (GRAS) status [1]; therefore it is a safe producer of many proteins and other compounds such as polyols, lipids and organic acids [9,10,11,12,13,14,15]. This oleaginous yeast is able to utilize many unspecific carbon sources such as fatty acids, alkanes or crude glycerol [16]. Crude glycerol, the main co-product of diesel production, contains many undesired contaminants such as methanol, salts or heavy metals; hence its market value is still relatively low [17]. However, despite the contaminations, Y. lipolytica can easily utilize this substrate [9].
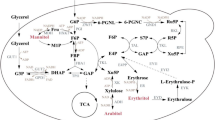
In yeast erythritol is produced as an osmoprotectant via the pentose phosphate pathway (PPP). Recently was shown, that also in Y. lipolytica its occurs via PPP [18]. The final step is catalyzed by erythrose reductase (ER), which reduces erythrose to erythritol with concomitant NAD(P)H oxidation [3, 19]. Several studies have been conducted on ER derived from Candida magnoliae [20], Moniliella megachiliensis (Trichosporonoides megachiliensis) [21] and Trichoderma reesei [22]. Moreover, it was shown that ER is a crucial gene in erythritol synthesis [4]. Despite this fact, the role of this enzyme in erythritol synthesis in Y. lipolytica is still unknown.
In this study we identified by in silico analysis predicted Y. lipolytica erythrose reductase (hereafter referred to as YlER), then we overexpressed the protein in Y. lipolytica and tested its influence on erythritol synthesis. In this study, we used glycerol as the sole carbon source for erythritol synthesis by Y. lipolytica, as it is a low-cost substrate that is beneficial for the industry. Moreover, we constructed a fusion of the YALI0F18590g gene encoding the hypothetical YlER with a C-terminal histidine tag to purify and characterize in enzymatic assays the protein for better understanding of its function. Here we found that overexpression of YALI0F18590g results in enhanced erythritol synthesis. Moreover, Zn2+ was found to be an important element for activity of YlER.
Methods
Microorganisms, media and culture conditions
The Y. lipolytica strains used in this study were derived from the wild-type Y. lipolytica A101 [23] and MK1 [19]. All of the strains used in this study are listed in Table 1.
Escherichia coli strains were cultivated in LB medium according to standard protocols [24]. Rich yeast extract peptone glucose (YPD) medium was used for the yeast inoculum preparation and protein overexpression and contained 1% (w/v) yeast extract, 1% (w/v) peptone and 2% (w/v) glucose.
During shake-flask experiments the cultures were grown in 0.3 l flasks containing 0.03 l medium on a rotary shaker (CERTOMAT IS, Sartorius Stedim Biotech) at 28 °C at 240 rpm. Erythritol synthesis was conducted in the following medium (g/L): 100 glycerol (Chempur, Poland), 2.3 (NH4)2SO4 (Chempur), 1 MgSO4 × 7H2O (Chempur), 0.23 KH2PO4 (Chempur), NaCl 26.4, 1 yeast extract (Merk, Germany) and 3 CaCO3, pH 3.0, supplemented with 0.25 mM ZnSO4 × 7H2O (Chempur), when required.
Bioreactor studies
To prepare an inoculation culture for fermentation in a bioreactor, the cultures were grown in 0.3 L flasks (containing 0.1 L of YPD medium) on a shaker at 28 °C for 72 h at 140 rpm. Erythritol production was conducted in a medium consisting of (g/L): 150 glycerol, 2.3 (NH4)2SO4, 1 MgSO4 × 7H2O, 0.23 KH2PO4, NaCl 26.4, 1 yeast extract, 0.25 mM ZnSO4 × 7H2O, pH 3.0.
Sequence analysis
The searches for protein sequence were performed with BLAST at the National Center for Biotechnology Information (http://blast.ncbi.nlm.nih.gov/Blast). The amino acid sequences of ER were aligned with aldo–keto reductase sequences using Clustal Omega software [25]. A phylogenetic tree was constructed with SeaView software using the neighbor-joining method [26].
Cloning and transformation protocols
All restriction enzymes were purchased from FastDigest Thermo Scientific (USA), and all of the digestions were performed according to standard protocols. The PCR reactions were set up using recommended conditions and Phusion high-fidelity DNA polymerase (Thermo Scientific). The ligation reactions were performed for 10 min at room temperature using T4 DNA Ligase (Thermo Scientific). The gel extractions were performed using the Gel Out gel extraction kit purchased from A&A Biotechnology (Poland). The E. coli minipreps were performed using the Plasmid Mini Kit (A&A Biotechnology). Transformation of E. coli strains was performed using standard chemical protocols [24]. Genomic DNA (gDNA) was extracted from Y. lipolytica using the Genomic Mini AX Yeast Spin kit (A&A Biotechnology).
Construction of plasmids
After amplification of Y. lipolytica erythrose reductase encoded by YALI0F18590g with primers YlER-AscI-F (5′–CATGGCGCGCCATGGCAGGCGGACCCAC–3′) and YlER-PmlI-R (5′-GCGCACGTGATTTAAATGCTAGCTTACTTCTTCTGCTCAGCAAGGTA–3′), the 1006 bp PCR fragment was digested with AscI and PmlI and cloned into the corresponding sites of pAD-UTGut1 [27] to yield pAD-YlER.
Next, primers YlER-AscI-F and YlER-his-R (5′-GCGGCTAGCTTAATGGTGAT GGTGATGGTGACGCGTCTTCTTCTGCTCAGCAAGGTA–3′) amplified YlER with a his-tag on the C terminal end (his-tag sequence underlined), yielding a 1016 bp PCR product. Consequently, the GUT1 gene was gel-extracted from plasmid pADUTGut1 [27] with AscI/NheI, and the obtained PCR product was inserted into the appropriate sites, to produce pAD-YlERhis.
The plasmids were digested with MssI to create linear expression cassettes devoid of E. coli DNA and surrounded by Y. lipolytica rDNA for targeted integrations. Y. lipolytica was transformed according to the lithium acetate method described previously [28]. The transformants were plated out on selective media [29]. Auxotrophies were restored via excision using the Cre-lox recombinase system following transformation with the replicative plasmid pUB4-Cre1(JME547) [30].
Expression and purification
First, the obtained transformants of Y. lipolytica AJD carrying the overexpression cassette YlER-his were grown in liquid YPD medium. Next, 4 × 100 mL of fresh YPD medium was inoculated to obtain starting OD600 at 0.4. Since YlER was cloned under the UAS1B16-TEF promoter, whose activity reached the highest level after 24 h of growth [31], growth of the strain was continued for the next 48 h. Then the recombinant strains were harvested by centrifugation at 4 °C for 10 min at 2500 rpm. After washing with phosphate-buffered saline (PBS, 10 mM phosphate, 150 mM NaCl, pH 7.4), harvested cells were resuspended in 5 vol of homogenization buffer containing 50 mM sodium phosphate buffer (pH 7.0), 300 mM NaCl and protease inhibitor cocktail (Sigma-Aldrich). The sample was sonicated on ice for 10 min and centrifuged for 30 min at 20,000 rpm at 4 °C. The his-tagged erythrose reductase proteins were purified by metal-chelated affinity chromatography using a 1 mL HisTALON Gravity Column (Clontech, USA) with 20 mM imidazole in the washing buffer and 150 mM imidazole in the elution buffer. The protein concentration was determined according to the Bradford method [32]. The purity of the final products was analyzed by SDS-PAGE and Western blotting with the anti-His (C-term)-AP antibody (R932-25, Invitrogen) according to standard protocols.
Enzyme assay
The enzymatic activity was determined as reported previously [4] with modifications given below. The reaction was initiated by mixing 1000 μL of 50 mM glycine–HCl/citrate/phosphate/Tris–HCl buffer (pH 2.5–9.0) containing 12 mM d-erythrose and 4 mM NADH and 10 μL of the purified enzyme. The absorbance at 340 nm was monitored at 22 °C for 10 min. The influence of metal ions on the activity of erythrose reductase was studied by assaying the enzyme activity at different ZnSO4, CuSO4, MnSO4 and FeSO4 concentrations (from 0.05 to 0.5 mM) at 28 °C in 50 mM citrate buffer (pH 3.0). For the effect of temperature, the activity of erythrose reductase was determined at different temperatures at pH 3.0. Substrate screening was performed with 4 mM NADH and 12 mM of the substrates l-arabinose, d-erythrose, d-fructose, d-glucose and d-galactose at a temperature of 28 °C at pH 3.0. The average of three measurements for each sample was adopted. One unit of enzyme activity was defined as the amount of enzyme which produced 1 μmol of NAD+ per minute under the above conditions.
Circular dichroism spectroscopy
Circular dichroism (CD) measurements were recorded on a Jasco model J-1500 spectropolarimeter equipped with a thermostated cell holder (JASCO, Tokyo, Japan). Experiments were performed in a quartz cell with a 5 mm path length over the range of 200–260 nm at various temperatures, pH and different concentrations of metal ions. During CD spectroscopy analysis, respective purified erythrose reductase (0.1 mg/mL) was resuspended in the corresponding buffer (pH 2.5–9.0) and analyzed at 28 °C. Next, the spectra were acquired at pH 3.0 and pH 7.5 with different temperatures (10–53 °C), respectively. CD spectra were collected with a data pitch of 0.1 nm, bandwidth of 2.0 nm and scanning speed of 50 nm/min. The spectra represent the average of 6–9 scans, and data were analyzed using the K2D3 method. The K2D3 web server is an online tool used to assess the secondary structural elements in the form of α-helices and β-strands from far-UV CD spectra ranging from 200 to 240 nm [33].
Fluorescence measurements
A cary eclipse fluorescence spectrophotometer was used for fluorescence measurements with an excitation wavelength of 280 nm, and the fluorescence emission was recorded in the range of 300–400 nm. The excitation and emission slit width (each 5 nm) was kept constant for all experiments. Fluorescence quenching measurements were recorded for erythrose reductase (15 mM) titrated with increasing concentrations of ZnSO4, CuSO4, MnSO4 and FeSO4 (0–0.5 mM) at pH 3.0 and room temperature (22 °C). All spectroscopic experiments were repeated at least three times.
Zeta-potential measurements
The measurements of the zeta-potential of the aqueous suspensions of erythrose reductase were carried out on a Malvern Zetasizer Nano-ZS analyzer (Malvern Instruments Ltd., Malvern, Worcestershire, UK). The pH titration was performed manually by starting with the native sample at pH 6.0, then changing the pH using 0.1 N HCl. Next a fresh sample was created and the pH was changed using 0.1 N NaOH. Three zeta potential measurements were made at each pH value, and the average value was reported.
Results and discussion
Yarrowia lipolytica gene identification
The ability of Y. lipolytica to produce erythritol has been known for many years [8, 34]. However, for this species the metabolic pathway of this phenomenon has never been described. It was shown that one of the crucial enzymes in erythritol synthesis is erythrose reductase [4], which catalyzes the final step in this process. Up to now, the erythrose reductase from Y. lipolytica has not been characterized. Based on our previous study [19], we selected eight predicted proteins from the aldo–keto reductase (AKR) superfamily and compared them with other erythrose reductases available from the NCBI database using the BLAST program. Among the chosen proteins, the one (XP 505585) encoded by the YALI0F18590g gene was characterized by the highest homology to C. magnoliae ER (ACT78580 CmER) (41% identity) and to three erythrose reductases of M. megachiliensis (BAD90687 MmER1, BAD90688 MmER2, BAD90689 MmER3) with 44% identity (see Additional file 1: Table S1). As mentioned above, these species are known for their high erythritol synthesis, and their ERs have been well described. These in silico data suggest that the protein encoded by YALI0F18590g (YlER) might play a similar role in Y. lipolytica. To verify BLAST analysis, we constructed a phylogenetic tree with full length amino acid sequences of selected AKRs from Y. lipolytica and other erythrose reductases using Clustal Omega software (Fig. 1). As seen in Fig. 1, only YlER (XP 505585) was close to the CmER and all three isoenzymes from M. megachiliensis. Additionally, this protein was located far from other AKRs from Y. lipolytica, suggesting that it might have a different role in the metabolism. Moreover, there is a possibility that YlER evolved differently for the other AKRs from Y. lipolytica. Given this result, we overexpressed the predicted erythrose reductase in Y. lipolytica to verify its involvement in the erythritol pathway.

Phylogenetic analysis of the aldo–keto reductases from Y. lipolytica. The phylogenetic tree was constructed based on the alignment of full sequences of amino acids of known erythrose reductases. Proteins are identified by their GenBank accession number
Effect of YlER overexpression on erythritol synthesis
The regulation and function of YlER protein is unknown in Y. lipolytica. Therefore, to verify the activity of all members of AKR family during erythritol synthesis, we compared the relative expression of the genes encoding the mentioned proteins. Remarkably, immediately after 24 h gene YALI0F18590g encoding YlER exhibited a 13-fold increase in expression (see Additional File 2: Figure S1A). Next, we overexpressed (see Additional File 2: Figure S1B) the predicted erythrose reductase to investigate whether the level of erythritol production was enhanced. For this purpose, the YALI0F18590g gene was cloned behind the constitutive UAS1B16-TEF promoter [31], the resulting plasmid pAD-YlER was digested, and consequently the overexpression cassette was introduced into the strain AMM. The strain AMM is derived from the well-characterized MK1 strain [19] that possesses natural ability for efficient production of erythritol from glycerol. It was found that production of erythritol occurs in high osmotic pressure conditions [35], so we performed a shake-flask experiment to verify the capacity of the engineered strain for erythritol synthesis and glycerol utilization. As a control, the parental strain MK1 was used. Interestingly, as seen in Fig. 2a, glycerol was depleted much faster by the engineered strain than by the control. The YlER overexpressing strain was able to utilize 100 g/L of glycerol within 58 h, whereas the control assimilated only 78.56 g/L. It was reported previously that overexpression of GUT1 and GUT2 genes (involved in assimilation of glycerol into the cell) results in enhanced utilization of glycerol [27]. Indeed we observed increased glycerol assimilation, whereas in this study the expression of GUT1 and GUT2 genes was not modified. However, the phenomenon of rapid glycerol assimilation was caused by pulling the metabolism toward erythritol synthesis [36] by overexpression of the last gene involved in this pathway. The biomass production for both of the strains remained at about the same level (12.7–12.85 g/L).

a Glycerol assimilation by AMM pAD-YlER (gray) and MK1 (black). Erythritol synthesis by AMM pAD-YlER (dark brown) and control strain (light brown); b erythritol synthesis in medium supplemented with 0.25 mM ZnSO4x7H2O by AMM pAD-YlER (gray bars) and control strain (black bars). The cultures were performed in triplicate. The error bars represent the standard deviation; c time courses of the 2 L scale batch fermentation in medium supplemented with 0.25 mM ZnSO4x7H2O glycerol assimilation by AMM pAD-YlER (gray) and control strain (black), erythritol synthesis by AMM pAD-YlER (dark brown) and control strain (light brown). The cultures were performed in triplicate. The error bars represent the standard deviation
In agreement with our assumptions, the engineered strain produced more erythritol than the control (Fig. 2a). The strain overexpressing YlER produced 44.4 g/L of erythritol, whereas the control (MK1) produced only 37.1 g/L. In addition, the process parameters such as erythritol productivity (QERY) and yield (YERY) were significantly enhanced. AMM pAD-YlER achieved QERY 0.77 g/L h, whereas MK1 achieved 0.64 g/L h; also YERY was enhanced and it reached 0.44 g/g, while YERY for the control was 0.37 g/g. This result is in agreement with a previous study, where the MK1 strain achieved 38.4 g/L of erythritol and YERY was 0.39 g/g [19]. Interestingly, recently it was observed that some strains of the Yarrowia clade possess the capacity for erythritol production [37]. However, in that study the highest titer of erythritol obtained by Yarrowia divulgata and Candida hollandica in a shake-flask experiment was below 40 g/L. Moreover, the strain Yarrowia lipolytica A101 produced less than 25 g/L of erythritol during 6 days of cultivation. So far, this is the first reported study employing metabolic engineering of the native gene involved in the erythritol pathway to enhance the titer of the desired polyol. Recently a published work by Woude et al. described heterologous expression of various ER in the cyanobacterium Synechocystis [38]. In that study the maximum titer of erythritol was obtained by strain SEP024, which produced only 256 mg/L of erythritol.
Here, the elevated level of erythritol synthesis in the engineered strain suggests that the gene YALI0F18590g is involved in metabolism of this polyol. In this study, we observed that production of erythritol increased by 20%, but probably it could be even more elevated. As mentioned above, AKRs require NAD(P)H as a cofactor to catalyze the reaction. The main pool of NAD(P)H is produced via PPP, but in this study we did not overexpress genes involved in this pathway. For this reason, part of the overexpressed protein remained inactive due to titration loss of the available cofactor. The excess of protein is not functional when the concentration of cofactor is not sufficient. This issue needs further studies for a fuller understanding. As the research field is interesting, further studies will be conducted on co-expression in the near future in our laboratory.
Next, we deleted YALI0F18590g to check its influence on erythritol synthesis. Deletion of YlER results in lower erythritol production compared to the control, however, the process is maintained (see Additional File 3: Figure S2). Probably the role of the YlER was intercepted by the other protein homologue(s).
Overexpression and purification of YlER
The next aim of our study was to investigate the biochemical properties of YlER. To overexpress YlER in Y. lipolytica, a plasmid harboring a fusion of the C-terminus of YlER with a his-tag was transformed into the AJD strain, resulting in AJD pAD-YlERhis. In this experiment, we chose the AJD strain that is derived from the Y. lipolytica A101 strain, because it is characterized by high biomass production [29]. Next, the engineered strain was grown for 48 h in YPD medium to obtain a high yield of the protein. To verify the overexpression of YlERhis following UASB16-TEF activity, SDS-PAGE and Western blot analysis were performed using his-tag specific antibodies (Fig. 3). The final yield of YlER calculated from the optical density at λmax was 1.6 mg/L of Y. lipolytica cells with a molecular mass of ~37 kDa, which corresponds well to the calculated mass and the range of aldo–keto reductases [39]. Finally, the YlER showed that its purity was >98% (Fig. 3). The experiment above demonstrates that a reasonably high yield of UASB16-TEF-dependent overexpression of YlER can be attained in Y. lipolytica. The purified protein was used for the further analysis.

a SDS-PAGE and Western-Blot b analysis of YlER-his. Lane M—protein standard, lane 1—purified YlER protein; lane 2—wash II; lane 3—crude cell extract
Biochemical properties of YlER
To examine substrate specificity, the protein activity was tested against various substrates. The results under standard assay conditions of pH 3.0 and 28 °C are presented in Table 2. The YlER was active with various aldose substrates. The enzyme showed a various specificity, with the highest specific activity against d-erythrose (6.51 U/mg), and lower specific activity against d-arabinose (6.25 U/mg) and d-galactose (6.11 U/mg). These obtained values were consistent with another characterized erythrose reductase enzyme from C. magnoliae [20] (Additional File 4: Table S2). To obtain more insight into the effect of pH and temperature on YlER activity we tested the activity of this protein in a wide range. First, the activity was tested at pH ranging from 2.5 to 9.0. As seen in Fig. 4a, the optimal pH for YlER activity is 3.0, at higher pH, its activity significantly decreases, and only about 15% of maximal activity was noted at pH above 4.0. This result explains the phenomenon of high titer of erythritol under acidic conditions. Previously, it has been observed that Y. lipolytica produces erythritol at low pH [8, 19] and during increasing pH of the environment the titer of erythritol decreases [40]. We suggest that this effect might be caused by inactivation of YlER. Higher pH of the environment results in carbon flux being pushed toward citric acid synthesis [12]. Given these results, we investigated the influence of temperature on YlER activity at pH 3.0. The maximum YlER activity was at 37 °C and slightly lower activity was noted at 28 °C. Below and above these temperatures the activity of YlER was significantly lower (Fig. 3b). Despite the fact that the optimal temperature for Y. lipolytica growth is 28–32 °C [1], YlER was the most active at 37 °C, this effect presumably being caused by higher kinetic energy of the molecules. Figure 4c presents the dependence of zeta-potential on pH for the analyzed YlER. At low pH, this protein presented positive zeta potentials, which decreased when pH was raised. The zeta-potential of YlER decreased from 23.5 mV at pH 2.5 to −33 mV at pH 8.5. The value of the isoelectric point (IEP), at which the zeta-potential of the enzyme was zero, was determined from Fig. 4c. The IEP of YlER was attained for pH 5.9. From theoretical calculations, the IEP of YlER was predicted to be 5.8. Our experimental IEP is slightly higher than the theoretical estimation, which can be explained by the fact that a small part of the charged amino acids is accessible to water.

a Effect of pH on specific activity of YlER; b temperature dependence of YlER activity; c functions of zeta-potential on pH for aqueous suspensions of erythrose reductase. The analysis was performed in triplicate. The error bars represent the standard deviation
Circular dichroism spectroscopy analysis of YlER
Next, the secondary structure of YlER was investigated by circular dichroism (CD) spectroscopy. The CD spectrum was measured by monitoring the changes of the signal from 200 to 260 nm. The CD spectrum (Fig. 5a) showed the shape of the mainly α-helical secondary structure of the negative ellipticity bands near 207 nm. The CD spectrum of YlER is very similar in shape to that of C. magnoliae erythrose reductase [20] and human aldose reductase [41], indicating that all proteins have similar folding patterns. To estimate the secondary structure composition, the spectra (with different molar extinction coefficients, Δε) were analyzed by the K2D3 method [33]. Fitting the spectrum of native erythrose reductase at pH 3.0 and temperature 28 °C yielded a 45.3% α-helical structure. The α-helix content of YlER was more than that in human aldose reductase (40% α-helix, PDB accession code, 2R24) [42]. This difference might explain the extent of the YlER function compared to other aldose reductases. The effect of pH on the secondary structure of YlER was analyzed at 28 °C. Figure 5a shows the changes of secondary structure at each pH point comparing within a native sample at pH 7.0. The results show that the shape of the spectrum is changed, which could be interpreted as the occurrence of conformational modifications. The secondary structure of YlER reveals a decrease in α-helical content from pH 7.5 to pH 9.0. In contrast, the α-helical content was found to increase from pH 7.0 to pH 2.5 (Table 3). Regarding YlER activity as a function of pH, the enzyme showed the highest activity at pH 3.0 (Fig. 4a). Moreover, at pH 3.0 and temperature 28 °C YlER had the highest α-helix structure (Fig. 5a). To the best of our knowledge, α-helix structures are implicated in aldose reductases folding [43] as an appropriate structure for catalyzes the NADH-dependent conversion activity. Figure 5b presents the CD spectra of the YlER, at the temperature between 10 and 53 °C at pH 3.0 and pH 7.5 (B and C, respectively). Interestingly, in the case of YlER, all the CD spectroscopy curves at different temperatures showed a similar pattern at pH 3.0 (Fig. 5b) and pH 7.5 (Fig. 5c). Deconvolution of data between 200 and 260 nm indicated that the content of α-helices was stable over a wide temperature range. Increasing from 28 to 53 °C caused YlER to lose about 1% at pH 3.0 and 2% at pH 7.5 of the α-helix content, respectively. The effect of temperature on the secondary structure indicated that the secondary structure of YlER is not sensitive to the environment. In our opinion, the structure of YlER was stabilized by the hydrogen bond, electrostatic interaction and the hydrophobic effect.

a Circular dichroism (CD) spectra of YlER at selected pH values and temperature 22 °C. Temperature dependence of circular dichroism (CD) spectra of YlER in 50 mM citrate/phosphate buffer at pH 3.0 (b) and pH 7.5 (c). The temperature ranges from 10 to 53 °C
Effects of divalent metal ions on YlER activity and stability
Previously, it has been shown that minerals can bind directly to enzymes and enhance their activity. Motivated by the fact that mineral supplementation enhances erythritol synthesis by many various microorganisms [21, 44, 45] we set out to determine whether minerals have an effect on YlER activity. The optimum pH of 3.0 as the most favorable condition was selected for analyzing the effects of metal ions on YlER activity. As shown in Table 4, addition of Zn2+ increases the activity of this protein. The activity increases by 5% at lower Zn2+ concentration (0.05 mM) and 10% at higher Zn2+ concentration (0.25 mM) at 28 °C. Several other metal ions including Cu2+, Mn2+ and Fe2+ also considerably inhibited the enzyme. Collectively, these results suggest that the enzyme has a metal-binding site that it can accept a wide variety of divalent metal ions, modulating the activity of YlER.
Next, in our study, we employed a fluorescence spectroscopy to evaluate the integrity of the protein structure. As YlER contains tryptophan (Trp) and tyrosine (Tyr) residues, its fluorescence emission by excitation at 280 nm should be dominated by fluorescence of Trp residues due to the intramolecular energy transfer from Tyr to Trp residues [46]. To explore the metal ion-induced fluorescence change of YlER, fluorescence measurements were performed at an excitation wavelength of 280 nm. Interestingly, we observed that various metal ions have a different influence on the fluorescence intensity of this protein. As shown in Fig. 6, addition of Cu2+, Fe2+ or Mn2+ significantly decreases the fluorescence intensity of the protein. Metal ion-induced fluorescence quenching of YlER is attributed to the complex formation between protein and metal ions, and this binding perturbs the microenvironment around the Trp and Tyr residues and causes fluorescence quenching of the protein. Interestingly, the addition of increasing concentrations of Zn2+ to YlER showed a progressive fluorescence increase of the Trp donor, thus decreasing resonance energy transfer efficiency. The maximum excitation and emission of YlER were at 280 and 334 nm, respectively. No obvious shift of the maximum emission wavelength of YlER was observed, indicating that divalent metal ions have no significant influence on the polarity of the Trp residue. The previous experiment showed that the presence of divalent metal ions might have an influence on YlER; therefore to monitor the secondary structure changes of YlER under the same conditions the CD measurement was performed. The spectra were scanned over the wavelength range of 200–260 nm. Figure 7 presents the CD spectra of YlER with the divalent cations under study. With the increase of Zn2+, Cu2+, Mn2+ and Fe2+ concentrations, the shape of the spectrum changed, which might be interpreted by the occurrence of conformational modifications. Mn2+ binding induces small decreases in secondary structure content, but upon Cu2+ and Fe2+ binding the small decreases in α-helical content are larger (Table 5). On the other hand, Zn2+ binding has the opposite effect observed for Cu2+, Mn2+ and Fe2+, as it significantly increases the secondary structure of YlER. Interestingly, Zn2+ increases its α-helix content from 45.3 to 46.8%. Moreover, Zn2+ has also been reported to be the activator of several enzymes, including carbonyl reductase [47], xylanase [48], nattokinase [49] and lipase [50]. The tendency for a difference between Zn2+ and other metal ions may be due to the different structural compatibility of metal ions to bind with YlER.

Fluorescence spectra of YlER in presence of increasing concentrations (0–0.5 mM) of Zn2+ (a), Cu2+ (b), Mn2+ (c) and Fe2+ (d)

Circular dichroism (CD) spectra of YlER at different concentrations of Zn2+ (a), Cu2+ (b), Mn2+ (c) and Fe2+ (d) between 0.05 and 0.5 mM
Effect of Zn2+ on erythritol production
Subsequently, we set out to determine whether Zn2+ supplementation increases erythritol synthesis by the engineered Y. lipolytica. In this experiment we compared AMM pAD-YlER strain harboring the overexpression cassette with its parental strain MK1. The strains were grown in baffled flasks in medium developed for erythritol synthesis, supplemented with 0.25 mM ZnSO4 × 7H2O. Interestingly, we noted a strong increase in erythritol titer for both of the strains, the engineered and the control strain (Fig. 2b). The erythritol titer was improved by 22 and 37% for the engineered (54.1 g/L) and the control strain (51.0 g/L), respectively, when compared to the results obtained in medium without zinc supplementation (Fig. 2a). In addition, for the engineered strain we noted an increase in YERY, which achieved 0.55 g/L/h. Interestingly, a significant difference was observed for the control, which achieved an erythritol titer (51.0 g/L) at a similar level to the engineered strain (44.4 g/L) in medium without supplementation. These results confirm that zinc has a great impact on production of this polyol by Y. lipolytica. Previously, it has been shown that zinc has a positive influence on erythritol synthesis [45]. However in that study, in medium supplemented with zinc the highest level of erythritol was 27.2 g/L. Probably the lower titer of polyol production was caused by lower osmotic pressure in the medium (medium without NaCl) and low rotation speed (140 rpm), which resulted in lower oxygenation of the medium. It has been noted before that different metal ions have a large impact on erythritol synthesis by various microorganisms [44, 51]. However, Zn2+ ions improved its production only in C. magnoliae and not in Torula sp. Moreover, the influence of metal ions on erythrose reductase activity has not been studied.
Next, to further characterize the engineered strain and explore its production abilities in medium supplemented with Zn2+ we performed large scale fermentation using a 5-L stirred-tank fermenter (Fig. 2c). Again, we used MK1 strain, as a control. In this experiment, the control strain produced 68.2 g/L of erythritol within 78 h of cultivation, with QERY 0.87 ± 0.05 g/L/h and YERY 0.45 ± 0.02 g/g. The strain overexpressing YlER produced 78.1 g/L of erythritol, and QERY and YERY were enhanced to 1.00 ± 0.12 g/L/h and 0.52 ± 0.06 g/g, respectively. Again, we observed enhanced erythritol synthesis by the engineered strain, nearly a 15% increase over the control bioreactor. It is important to note that also the process parameters (such as yield and productivity) were enhanced, what is beneficial for process development.
In summary, these experiments showed that Zn2+ has a positive influence on YlER activity and is an important factor in erythritol synthesis by Y. lipolytica. Moreover, it indicates the importance of environmental conditions on genetic targets for metabolic engineering.
Conclusions
In this study we found that the predicted protein from the aldo–keto reductase (AKR) superfamily encoded by the YALI0F18590g gene is an erythrose reductase, which plays an important role in erythritol synthesis in Y. lipolytica. To our knowledge, this is the first reported efficient production of erythritol by genetically modified microorganisms by overexpression of the native ER. Moreover, the overexpression of YlER coupled with Zn2+ supplementation results in robust erythritol production from glycerol. This enzyme can be heterologously expressed in microorganisms which do not synthesize erythritol such as cyanobacteria. Further studies will focus on metabolic engineering, leading to an elevated NAD(P)H pool and optimization of culture conditions to improve the erythritol production capacities of Y. lipolytica.
Abbreviations
- YlER:
-
Y. lipolytica gene YALI0F18590g
- ARK:
-
aldo–keto reductase superfamily
- ERY:
-
erythritol
- Q:
-
productivity
- Y:
-
yield
References
Groenewald M, et al. Yarrowia lipolytica: safety assessment of an oleaginous yeast with a great industrial potential. Crit Rev Microbiol. 2014;40(3):187–206.
Noda K, Nakayama K, Oku T. Serum glucose and insulin levels and erythritol balance after oral administration of erythritol in healthy subjects. Eur J Clin Nutr. 1994;48(4):286–92.
Moon HJ, et al. Biotechnological production of erythritol and its applications. Appl Microbiol Biotechnol. 2010;86(4):1017–25.
Lee DH, et al. Molecular cloning and biochemical characterization of a novel erythrose reductase from Candida magnoliae JH110. Microb Cell Fact. 2010;9:43.
Park J, et al. Production of erythritol in fed-batch cultures of Trichosporon sp. J Ferment Bioeng. 1998;86(6):577–80.
Kohl ES, et al. Scale-up of erythritol production by an osmophilic mutant of Candida magnoliae. Biotechnol Lett. 2003;25(24):2103–5.
Segueilha L. Method producing erythritol by repeated fed-batch fermentation, in 09/337909 United States Patent. Freres R, Editor. 2002.
Rymowicz W, Rywinska A, Marcinkiewicz M. High-yield production of erythritol from raw glycerol in fed-batch cultures of Yarrowia lipolytica. Biotechnol Lett. 2009;31(3):377–80.
Dobrowolski A, et al. Efficient conversion of crude glycerol from various industrial wastes into single cell oil by yeast Yarrowia lipolytica. Bioresour Technol. 2016;207:237–43.
Kamzolova SV, et al. Production of technical-grade sodium citrate from glycerol-containing biodiesel waste by Yarrowia lipolytica. Bioresour Technol. 2015;193:250–5.
Morgunov IG, Kamzolova SV, Lunina JN. The citric acid production from raw glycerol by Yarrowia lipolytica yeast and its regulation. Appl Microbiol Biotechnol. 2013;97(16):7387–97.
Papanikolaou S, et al. Yarrowia lipolytica as a potential producer of citric acid from raw glycerol. J Appl Microbiol. 2002;92(4):737–44.
Rakicka M, et al. Lipid production by the oleaginous yeast Yarrowia lipolytica using industrial by-products under different culture conditions. Biotechnol Biofuels. 2015;8:104.
Rymowicz W, et al. Citric acid production from glycerol-containing waste of biodiesel industry by Yarrowia lipolytica in batch, repeated batch, and cell recycle regimes. Appl Microbiol Biotechnol. 2010;87(3):971–9.
Lazar Z, et al. Optimized invertase expression and secretion cassette for improving Yarrowia lipolytica growth on sucrose for industrial applications. J Ind Microbiol Biotechnol. 2013;40(11):1273–83.
Zhu Q, Jackson EN. Metabolic engineering of Yarrowia lipolytica for industrial applications. Curr Opin Biotechnol. 2015;36:65–72.
Makri A, Fakas S, Aggelis G. Metabolic activities of biotechnological interest in Yarrowia lipolytica grown on glycerol in repeated batch cultures. Bioresour Technol. 2010;101(7):2351–8.
Mirończuk AM, Biegalska A, Dobrowolski A. Functional overexpression of genes involved in erythritol synthesis in the yeast Yarrowia lipolytica. Biotechnol Biofuels. 2017;10(1):77.
Mirończuk AM, et al. Newly isolated mutant of Yarrowia lipolytica MK1 as a proper host for efficient erythritol biosynthesis from glycerol. Process Biochem. 2015;50(1):61–8.
Lee JK, et al. Purification and characterization of a novel erythrose reductase from Candida magnoliae. Appl Environ Microbiol. 2003;69(7):3710–8.
Ookura T, et al. Primary structure analysis and functional expression of erythrose reductases from erythritol-producing fungi (Trichosporonoides megachiliensis SNG-42). Biosci Biotechnol Biochem. 2005;69(5):944–51.
Jovanovic B, Mach RL, Mach-Aigner AR. Characterization of erythrose reductases from filamentous fungi. AMB Express. 2013;3(1):43.
Wojtatowicz M, Rymowicz W. Effect of inoculum on kinetics and yield of citric acids production on glucose by Yarrowia lipolytica A-101. Acta Aliment Pol (Poland). 1991;41(2):137–43.
Sambrook J, Russell D. Molecular cloning: a laboratory manual. 3rd ed. New York: Cold Spring Harbor Laboratory Press; 2001.
Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol. 2011;7:539.
Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27(2):221–4.
Mirończuk AM, et al. A novel strain of Yarrowia lipolytica as a platform for value-added product synthesis from glycerol. Biotechnol Biofuels. 2016;9(1):1–12.
Nicaud JM, Fabre E, Gaillardin C. Expression of invertase activity in Yarrowia lipolytica and its use as a selective marker. Curr Genet. 1989;16(4):253–60.
Mirończuk AM, et al. A two-stage fermentation process of erythritol production by yeast Y. lipolytica from molasses and glycerol. Bioresour Technol. 2015;198:445–55.
Fickers P, et al. New disruption cassettes for rapid gene disruption and marker rescue in the yeast Yarrowia lipolytica. J Microbiol Methods. 2003;55(3):727–37.
Blazeck J, et al. Tuning gene expression in Yarrowia lipolytica by a hybrid promoter approach. Appl Environ Microbiol. 2011;77(22):7905–14.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
Louis-Jeune C, Andrade-Navarro MA, Perez-Iratxeta C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins. 2012;80(2):374–81.
Abe, S, Morioka S. Method of producing erythritol. 1999, Google Patents.
Yang L-B, et al. A novel osmotic pressure control fed-batch fermentation strategy for improvement of erythritol production by Yarrowia lipolytica from glycerol. Biores Technol. 2014;151:120–7.
Tai M, Stephanopoulos G. Engineering the push and pull of lipid biosynthesis in oleaginous yeast Yarrowia lipolytica for biofuel production. Metab Eng. 2013;15:1–9.
Rakicka M, et al. Sweet and sour potential of yeast from the Yarrowia clade. Biomass Bioenerg. 2016;92:48–54.
van der Woude AD, et al. Genetic engineering of Synechocystis PCC6803 for the photoautotrophic production of the sweetener erythritol. Microb Cell Fact. 2016;15:60.
Barski OA, Tipparaju SM, Bhatnagar A. The aldo–keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev. 2008;40(4):553–624.
Tomaszewska L, et al. A comparative study on glycerol metabolism to erythritol and citric acid in Yarrowia lipolytica yeast cells. FEMS Yeast Res. 2014;14(6):966–76.
Petrash JM, et al. Kinetic alteration of human aldose reductase by mutagenesis of cysteine residues. Adv Exp Med Biol. 1993;328:289–300.
Blakeley MP, et al. Quantum model of catalysis based on a mobile proton revealed by subatomic X-ray and neutron diffraction studies of h-aldose reductase. Proc Natl Acad Sci USA. 2008;105(6):1844–8.
Borhani DW, Harter TM, Petrash JM. The crystal structure of the aldose reductase. NADPH binary complex. J Biol Chem. 1992;267(34):24841–7.
Lee J-K, et al. Increased erythritol production in Torula sp. by Mn2+ and Cu2+. Biotech Lett. 2000;22(12):983–6.
Tomaszewska L, Rymowicz W, Rywińska A. Mineral supplementation increases erythrose reductase activity in erythritol biosynthesis from glycerol by Yarrowia lipolytica. Appl Biochem Biotechnol. 2014;172:1–10.
Liu X, et al. Effects of metal ions and an inhibitor on the fluorescence and activity of acutolysin A from Agkistrodon acutus venom. Indian J Biochem Biophys. 2005;42(2):100–5.
Chen XH, et al. A novel carbonyl reductase with anti-Prelog stereospecificity from Acetobacter sp. CCTCC M209061: purification and characterization. PLoS ONE. 2014;9(4):e94543.
Fortkamp D, Knob A. High xylanase production by Trichoderma viride using pineapple peel as substrate and its application in pulp biobleaching. Afr J Biotechnol. 2014;13(22):2248–59.
Wang C, et al. Purification and characterization of nattokinase from Bacillus subtilis natto B-12. J Agric Food Chem. 2009;57(20):9722–9.
Prive F, et al. Identification and characterization of three novel lipases belonging to families II and V from Anaerovibrio lipolyticus 5ST. PLoS ONE. 2013;8(8):e69076.
Savergave LS, et al. Strain improvement and statistical media optimization for enhanced erythritol production with minimal by-products from Candida magnoliae mutant R23. Biochem Eng J. 2011;55(2):92–100.
Hanahan D, Glover DM. DNA cloning: a practical approach, vol. 1. Virginia: IRL Press, McLean; 1985. p. 109.
Authors’ contributions
TJ purified the protein, performed biochemical analysis, circular dichroism spectroscopy analysis. AB participated in the fermentation experiments. AD performed in silico analysis and revised the manuscript. AMM designed the study, constructed the plasmids and strains, participated in the fermentation experiments, and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Magdalena Rakicka for HPLC analysis.
Competing interests
The authors declare that they have no competing interests.
Availability of supporting data
The authors promise the availability of supporting data.
Consent for publication
The authors have consented for publication.
Funding
This work was financed by the Polish National Centre for Research and Development under Project LIDER/010/207/L-5/13/NCBR/2014.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
12934_2017_733_MOESM2_ESM.doc
Additional file 2: Figure S1. Quantification of genes expression belong to the ARK family, during erythritol synthesis (A). Quantification of YALI0F18590g gene expression (YlER,) by the strain AMM pAD-YIER and the control (B). Samples were analyzed in triplicate and the standard errors were estimated using Illumina Eco software. The results were normalized to actin gene ACT-F/ACT-R and analyzed using the ddCT method.
12934_2017_733_MOESM3_ESM.doc
Additional file 3: Figure S3. Erythritol synthesis by strain AMM ΔYlER (gray bars) and control strain MK1 (black bars). The cultures were performed in triplicate. The error bars represent the standard deviation.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Janek, T., Dobrowolski, A., Biegalska, A. et al. Characterization of erythrose reductase from Yarrowia lipolytica and its influence on erythritol synthesis. Microb Cell Fact 16, 118 (2017). https://doi.org/10.1186/s12934-017-0733-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-017-0733-6