
Abstract
Background
The implementation of novel chassis organisms to be used as microbial cell factories in industrial applications is an intensive research field. Lactococcus lactis, which is one of the most extensively studied model organisms, exhibits superior ability to be used as engineered host for fermentation of desirable products. However, few studies have reported about genome reduction of L. lactis as a clean background for functional genomic studies and a model chassis for desirable product fermentation.
Results
Four large nonessential DNA regions accounting for 2.83% in L. lactis NZ9000 (L. lactis 9 k) genome (2,530,294 bp) were deleted using the Cre-loxP deletion system as the first steps toward a minimized genome in this study. The mutants were compared with the parental strain in several physiological traits and evaluated as microbial cell factories for heterologous protein production (intracellular and secretory expression) with the red fluorescent protein (RFP) and the bacteriocin leucocin C (LecC) as reporters. The four mutants grew faster, yielded enhanced biomass, achieved increased adenosine triphosphate content, and diminished maintenance demands compared with the wild strain in the two media tested. In particular, L. lactis 9 k-4 with the largest deletion was identified as the optimum candidate host for recombinant protein production. With nisin induction, not only the transcriptional efficiency but also the production levels of the expressed reporters were approximately three- to fourfold improved compared with the wild strain. The expression of lecC gene controlled with strong constitutive promoters P5 and P8 in L. lactis 9 k-4 was also improved significantly.
Conclusions
The genome-streamlined L. lactis 9 k-4 outcompeted the parental strain in several physiological traits assessed. Moreover, L. lactis 9 k-4 exhibited good properties as platform organism for protein production. In future works, the genome of L. lactis will be maximally reduced by using our specific design to provide an even more clean background for functional genomics studies than L. lactis 9 k-4 constructed in this study. Furthermore, an improved background will be potentially available for use in biotechology.
Similar content being viewed by others
Background
Bacteria are commonly used as microbial cell factories for metabolic engineering and desirable product fermentation at the laboratory scale and in industrial applications [ 1, 2 ]. For Gram-negative bacteria, Escherichia coli is a key organism utilized to construct a genetically stable strain that demonstrates robust metabolic performance [3, 4]. Shen and co-workers achieved high-titer anaerobic 1-butanol synthesis in E. coli [5]. Moon and co-workers achieved production of glucaric acid from a synthetic pathway in recombinant E. coli [6]. Hashimoto and co-workers showed that the cell size and nucleoid organization of E. coli cells can be changed through genome reduction [7]. The minimized E. coli displayed some convenience as a host to express target products, but several disadvantages were observed, such as the formation of endotoxins and inclusion in intracellular protein production [8, 9]. Pseudomonas putida is another bacterium selected to be constructed as robust heterologous gene expression platform. The work of de Lorenzo and co-workers showed that the streamlined-genome derivatives of P. putida KT2440 out competed the parental strain in every industrially relevant trait assessed, and the mutants reached a recombinant protein yield with respect to biomass of up to 40% higher than that of the wild strain [10, 11]. For Gram-positive bacteria, Morimoto and co-workers reported that they deleted 874 kb (20%) of the genomic sequence in Bacillus subtilis MBG874, and the heterologous protein productivity was remarkably enhanced in the mutant [12]. Unthan and co-workers initiated the construction of a chassis from Corynebacterium glutamicum ATCC13032 by decreasing the size of the native genome. Five strains with combinatory deletions of irrelevant gene clusters were investigated (GRS22-23, 44.0 kb deleted, accounting to 1.34% of genome; GRS23-46, 215.9 kb deleted, 6.58%; GRS16-23, 165.2 kb deleted, 5.03%; GRS21-41, 215.2 kb deleted, 6.55%; GRS41-51, 108.7 kb deleted, 3.31%); among them, three potential candidates exist, namely, GRS22-23, GRS23-46, and GRS16-23, which can be used for chassis construction [13].
Lactococcus lactis, which is one of the most extensively studied model organisms, exhibits superior ability to be used as engineered host for fermentation of desirable products. Purification of the targeted products from L. lactis is very convenient because it does not produce any endotoxins, inclusions nor many unwanted products [14, 15]. Genome reduction of L. lactis, which is designated as a GRAS (Generally Regarded As Safe) organism, can be applicable to a model chassis for fermentation of desirable products. Many possibly identified nonessential large DNA regions should be deleted to provide an efficient background for use in biotechnology applications and a clean background for functional genomics studies.
However, few studies are available about genome reduction of L. lactis despite its importance. In previous reports, some genes, such as htrA, clpP, and ybdD, which were involved in heterologous protein production and secretion, have been analyzed in L. lactis. HtrA is an extracellular protease in L. lactis and B. subtilis, and this protease plays an important role in the degradation of heterologous proteins [16]. The production of heterologous proteins can be improved when HtrA is deleted. ClpP is an intracellular protease also affecting the production of heterologous proteins [17]. The ybdD mutant strain shows increased levels of exported proteins [18]. The multiple protease mutant strains were also reported in Laxmi’s work [19]. In his study, not only the degradation of heterologous protein was reduced, but also the levels of cell-associated protein-folding catalysts were elevated in the multiple protease mutants. The deletion of multiple protease genes in L. lactis and B. subtilis can be an important beneficial element in the construction of protein-secreting strains [20]. To date, the construction and use of genome-streamlined L. lactis as microbial cell factory remain as attractive alternative methods to improve protein expression.
In our study, four large nonessential DNA regions accounting for 2.83% of the genome, such as prophages, transposons, and related proteins, were selected and deleted with the Cre-loxP deletion system in L. lactis NZ9000 [21]. The mutants were compared with the wild strain in several physiological traits. The mutants were also evaluated as microbial cell factories for recombinant protein production (intracellular and secretory expression) with the red fluorescent protein (RFP) [22] and bacteriocin leucocin C (LecC) [23] as reporters controlled by the nisin inducible P nisZ and strong constitutive promoters P8 and P5 [24]. The genome-streamlined mutant L. lactis 9 k-4 outcompeted the wild strain in several physiological traits assessed. Additionally, L. lactis 9 k-4 exhibited the optimal properties among mutants as host for protein production.
Results
Design and construction of platform L. lactis strain for heterologous protein production
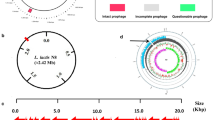
To construct the streamlined-genome mutants, we initially searched the prophage, prophage-like, and transposon genes in L. lactis 9 k genome sequences because prophages and transposons may not always be necessary for the strain cultured in the laboratory or industrial condition. Five prophages and related genes were identified. Four of them were successfully deleted step-by-step. The distribution of four deleted large DNA regions throughout the genome is indicated in Fig. 1a. The genetic organization of the four deleted DNA regions is shown in Fig. 1b. The first large nonessential DNA region-L1 (containing prophage 1 and related genes), which is similar to Staphylococcus aureus bacteriophages 80α, is 9.7 kb and contains 15 open reading frames (ORFs) [25]. Among these genes, seven have been characterized and one encodes integrase. Region L2 comprises 22.5 kb with 35 ORFs. Region L2 encodes one integrase, two transposase (IS712H)-related proteins, and five prophage ps1-related proteins. Region L3 exhibits a size of 17.9 kb and contains 34 ORFs. Eight repeat regions and six prophage-related proteins exist in L3. The phage in L3 was identified as phage Q33; Q33 is a member of the P335 species, and some of its genes encode functions such as DNA replication and packaging, morphogenesis, and host cell lysis [26]. Finally, region L4 comprises 21.6 kb, including 30 coding sequences that, except four of them, are all oriented in the same direction. Region L4 encodes 14 prophage-related proteins and three repeat regions. The phage was identified as ΦC31 through NCBI gene blast. The integrase of ΦC31 can integrate some exogenous plasmids carrying an attB site into native genomic sequences that bear partial identity to attP [27]. Detailed description of the genes is provided in Additional file 1: Table S1.

Genetic organization and deletion of four large nonessential DNA regions in L. lactis 9 k. a Circular map of L. lactis 9 k chromosome showing the physical location of the deletions in the genome; b genetic organization of four large deletions; c distribution and annotations of ORFs found in the large deletions
Overall, the deletion of DNA region contents of L. lactis 9 k comprises approximately 2.83% of its genome. Figure 1c shows the percentage of the total ORFs of four large deleted DNA regions grouped in a gross functional classification. Notably, the account reveals ∼54.4% of the corresponding ORFs encoding proteins of unknown function. The deletion results were verified by polymerase chain reaction (PCR) with testing primers (Additional file 1: Figure S1) and sequence analysis.
Assessment of mutants’ growth parameters
The growth profiles of strains were monitored by following the optical density at 600 nm (OD600) of cells in M17G and SA media. The growth curves are shown in Fig. 2a (M17G media) and Fig. 2b (SA media). As presented in Fig. 2a, the results showed that all the mutants started to grow exponentially 1 h before the parental strain. However, the lag growth phase of different strains exhibited no significant difference in M17G media. As shown in Fig. 2b, L. lactis 9 k-2, L. lactis 9 k-3, and L. lactis 9 k-4 started to grow exponentially 2 h before the wild strain when the strains were cultured in defined SA media. By contrast, L. lactis 9 k-1 grew similar to the wild strain. The final cell density of L. lactis 9 k-2 was approximately 1.2-fold higher than the wild strain, whereas the final cell densities of L. lactis 9 k-3 and L. lactis 9 k-4 were approximately 1.4-fold higher than that of the wild strain. The maximum specific growth rate (μmax, maximal slope of growth curve) of strains was determined during exponential growth. L. lactis 9 k-4 and L. lactis 9 k-3 showed 17 and 10% increase in μmax when grown in M17G media, respectively. No significant difference was observed among L. lactis 9 k-1, L. lactis 9 k-2, and the wild strain. Interestingly, L. lactis 9 k-2 showed a 19% increase and L. lactis 9 k-4 showed a 32% increase in μmax when grown in SA media. The μmax difference increased between the wild strain and mutants when the strains were cultured in defined SA media.

Growth profiles and maximum specific growth rate (μmax) of strains. The μmax was determined during exponential growth. a Cultured in M17G media; b cultured in SA media. Data represent the mean of three independent experiments. Bars indicate standard deviations (*P < 0.05)
All mutants attained increased final cell dry weight (CDW) on glucose (Yx/s, gCDW g −1glucose ) grown in M17G or SA media (Fig. 3a). The difference reached the maximum when the cells were cultured for 10 h in SA media. The biomass yield coefficient Y X/S of L. lactis 9 k-3 was approximately 0.28 showing a 7% increase, and L. lactis 9 k-4 was approximately 0.32 showing a 15% increase. The energetic capacity of the cells can be estimated with the amount of adenosine triphosphate (ATP) per unit of biomass (Y ATP/X, μmol g −1CDW ) [9]. Therefore, the corresponding ATP concentrations were measured. Moreover, the results were normalized to CDW to evaluate the energetic capacity of cells (Fig. 3b). The calculated results showed that Y ATP/X of all the mutants was higher than that of the parental strain, and attained the maximum difference in SA media when grown for 10 h. In addition, L. lactis 9 k-3 showed a 13% increase, and L. lactis 9 k-4 showed a 21% increase in Y ATP/X compared with the wild strain.

Biomass coefficient and yield of ATP on biomass. 1, L. lactis 9 k; 2, L. lactis 9 k-1; 3, L. lactis 9 k-2; 4, L. lactis 9 k-3; 5, L. lactis 9 k-4. a The biomass yield coefficient (Y X/S); b the yield of ATP on biomass (Y ATP/X). Data represent the mean of three independent experiments. Bars indicate standard deviations
Assessment of mutants’ phenotype
Extensive fermentation phenotype analyses of L. lactis 9 k-1, L. lactis 9 k-2, L. lactis 9 k-3, L. lactis 9 k-4, and L. lactis 9 k were conducted using the phenotype microarrays to understand further the physiological difference between the wild and mutant strains. All of the substrates that the mutant strains consumed were significantly different compared with the L. lactis 9 k, as summarized in Table 1. The results showed that L. lactis 9 k-2, L. lactis 9 k-3, and L. lactis 9 k-4 can efficiently metabolize nine carbon sources, particularly d-gluconic acid, maltose, d-fructose, α-d-glucose, and α-methyl-d-dgalactoside stachyose. The mutants metabolized approximately six carbon sources less efficiently. The largest deletion mutant L. lactis 9 k-4 exhibited good properties on some carbon source metabolism compared with the wild strain, especially for α-d-glucose and d-fructose.
Assessment of lecC gene expression in mutant strains
The expression of lecC gene was analyzed at transcriptional and production levels through quantitative reverse transcription polymerase chain reaction (RT-qPCR) and agar-diffusion experiment. The difference of leucocin C transcriptional and production efficiency of different hosts cultured in M17G or in SA media with or without nisin to induce expression was analyzed. The results showed that the transcriptional difference of the lecC gene achieved the maximum when the cells were cultured for 10 h with or without nisin induction (Fig. 4a, b). The transcription of the lecC gene in L. lactis 9 k-4-lecC, L. lactis 9 k-3-lecC, and L. lactis 9 k-2-lecC was approximately 3.1-, 2.5-, and 2.2-fold higher than that of the wild strain, respectively, when they were cultured in M17G media with or without nisin addition for 10 h (Fig. 4a). Moreover, L. lactis 9 k-4-lecC, L. lactis 9 k-3-lecC, and L. lactis 9 k-2-lecC achieved 4.1-fold, 3.3-fold, and 2.6-fold higher transcription level of the lecC gene than that of the wild strain, correspondingly, when the strains were cultured in SA medium (Fig. 4b).

Relative quantity of lecC and rfp mRNA. 1, L. lactis 9 k-lecC; 2, L. lactis 9 k-1-lecC; 3, L. lactis 9 k-2-lecC; 4, L. lactis 9 k-3-lecC; 5, L. lactis 9 k-4-lecC; a, L. lactis 9 k-lecC; b, L. lactis 9 k-4-P5/lecC; c, L. lactis 9 k-4-P8/lecC. a Cultured in M17G media; b cultured in SA media; c cultured in SA or in M17G media without nisin; d cultured in SA or in M17G media with nisin to induce expression. Data represent the mean of three independent experiments. Bars indicate standard deviations (*P < 0.05)
The results of antibacterial experiment (Fig. 5) showed that the inhibiting zones of L. lactis 9 k-2-lecC, L. lactis 9 k-3-lecC, and L. lactis 9 k-4-lecC were obviously larger than that of the wild strain L. lactis 9 k-lecC regardless whether they were cultured in M17G or in SA media. When the strains were cultured for 10 and 14 h, the inhibiting zones of L. lactis 9 k-4-lecC, L. lactis 9 k-3-lecC, and L. lactis 9 k-2-lecC were approximately 2.5-, 2.0-, and 1.5-fold larger than that of the wild strain, respectively, when they were induced with nisin to express lecC. Growth in M17G or SA media did not influence the fold differences. Without nisin induction, the lecC gene was transcribed from the constitutive P45 promoter resulting in inhibition zones of 1.5-, 1.3-, and 1.2-fold larger for the L. lactis 9 k-4-lecC, 9 k-3-lecC, and 9 k-2-lecC strains than those of the wild strain, respectively. No significant difference was observed in lecC expression between L. lactis 9 k-1-lecC and the wild strain.

Comparison of antibacterial activity of leucocin C expressed in different mutants. I induced expression, N non-induced expression. The commercial nisin were added to the cultures at a concentration of 10 IU/ml when needed
Assessment of lecC gene expression under control of strong constitutive promoters in L. lactis 9 k-4
The strong constitutive promoters P8 and P5 were selected from the L. lactis promoter library and cloned in front of the lecC gene for expression in L. lactis 9 k-4. As presented in Fig. 4c, the results showed that the transcriptional efficiency of the lecC gene was improved significantly when it was controlled under the strong constitutive promoters P8 and P5. The transcription of lecC gene was improved by approximately 5.7-fold with promoter P8 and 5.0-fold with promoter P5 when the hosts were cultured in SA media. The transcription of lecC gene was also improved by approximately 4.8-fold with promoter P8 and 4.1-fold with promoter P5 when the hosts were cultured in M17G media.
The inhibiting zones of L. lactis 9 k-4-P8/lecC and L. lactis 9 k-4-P5/lecC were obviously larger than that of L. lactis 9 k-lecC regardless whether they were cultured in M17G or SA media (Fig. 6). The inhibiting zones of L. lactis 9 k-4-P8/lecC were approximately 3.0-fold larger than that of the wild strain when the strains were cultured for 10 and 14 h. The inhibiting zones of L. lactis 9 k-4-P5/lecC were approximately 2.5-fold larger when they were non-induced to express lecC in M17G media. The inhibition zones of L. lactis 9 k-4-P8/lecC and L. lactis 9 k-4-P5/lecC were 2.5- and 2.0-fold larger than that of the wild strain in SA media, respectively.

Comparison of antibacterial activity of leucocin C under control of the strong constitutive promoters P5 and P8 in the L. lactis 9 k-4. N non-induced expression
Assessment of rfp gene expression in mutant strains
The expression of the rfp gene was analyzed at transcriptional and production levels through RT-qPCR and Infinite M200 plate reader system. The transcriptional efficiency of the rfp reporter gene of mutant strains cultured in M17G or SA media with nisin to induce expression was analyzed (Fig. 4d). The results showed that the transcriptional difference of the rfp gene reached the maximum when the cells were cultured for 10 h in SA or M17G media with an inducer. The transcription of the rfp gene in L. lactis 9 k-4-rfp was approximately 3.0-fold higher than that of the wild strain. By contrast, the transcription of the rfp gene of L. lactis 9 k-3-lecC was approximately 2.2-fold higher and that of L. lactis 9 k-2-lecC was approximately 2.1-fold higher than that of the wild strain when they were cultured in M17G media for 10 h. The transcription of the rfp gene achieved sequentially 3.7-, 2.9-, and 2.3-fold higher than that of the wild strain when the strains were cultured in SA media.
The fluorescence intensity was traced for 20 h with Infinite M200 plate reader. This intensity reached the maximum when the cells were cultured in a 96-well plate for 17 h. The maximum specific rate (Kmax = △RFP/△t) of RFP formation in different mutants was calculated during the exponential growth. As presented in Fig. 7, the results showed that the fluorescence intensity and Kmax of L. lactis 9 k-4-rfp and L. lactis 9 k-3-rfp were obviously higher than that of the wild strain L. lactis 9 k-lecC regardless whether they were cultured in M17G or SA media. As shown in Fig. 7a, the fluorescence intensity of L. lactis 9 k-4-rfp was approximately 2.2-fold stronger than that of the wild strain L. lactis 9 k-rfp. By contrast, the fluorescence intensity of L. lactis 9 k-3-rfp increased by almost 1.7-fold, and that of L. lactis 9 k-2-rfp increased approximately by 1.5-fold. At the same time, L. lactis 9 k-4 and L. lactis 9 k-3 showed a 27 and 18% increase in Kmax, respectively. No statistical significant difference was observed among L. lactis 9 k-1, L. lactis 9 k-2, and wild strain when the cells were cultured in M17G media. The difference in fluorescence intensity between wild and mutant strains became large when they were cultured in SA media. As shown in Fig. 7b, the fluorescence intensity of L. lactis 9 k-4-rfp was approximately 3.1-fold stronger than that of the wild strain, whereas the fluorescence intensity of L. lactis 9 k-3-rfp was approximately 2.2-fold. Moreover, L. lactis 9 k-4 showed a 15% increase and L. lactis 9 k-3 showed a 7% increase in Kmax.

Comparison of fluorescence intensity of RFP and maximum specific rate of RFP formation (Kmax) in different mutants. Kmax was determined during exponential growth. The strain containing plasmid pNZ8048 was used as control. a Cultured in M17G media; b cultured in SA media. Data represent the mean of three independent experiments. Bars indicate standard deviations (*P < 0.05)
Discussion
L. lactis NZ9000 has been frequently used as an expression host for nisin-induced protein production [28–30]. In this study, the genome of this host was reduced by four consecutive detetions of nonessential DNA regions with Cre-loxP deletion system (altogether 2.83% of the genome). The mutant strains were compared with the parental strain with regard to several physiological traits (μmax, Kmax, Y X/S, and Y ATP/X) and evaluated as microbial cell factories for heterologous protein production. The results showed that all mutants exhibited good characteristics in μmax, Y X/S, and Y ATP/X compared with the parental strain, especially L. lactis 9 k-4, which can produce more CDWs and available ATPs, as well as increased μmax with the same glucose consumption. Two proteins, RFP for cytoplasmic expression and bacteriocin LecC for secreted expression, were expressed in the deletion strains to test if the decreased metabolic burden would enable the host to produce efficiently heterologous proteins. These proteins were also compared with the expression in the wild strain. The results showed that decreasing the genome size yielded hosts with an increased capacity to express heterologous proteins into the cytoplasm and secreted to the medium. L. lactis 9 k-1 with the least decreased genome size presented the lowest effect on protein production. Increasing the genome deletion enhances the strain to perform as production host. This phenomenon was also reflected similarly in how well the cells grew in SA media and at the transcriptional level analyzed by RT-qPCR. In particular, L. lactis 9 k-4 with the largest deletions produced most RFP and LecC. Moreover, L. lactis 9 k-4 achieved the highest cell densities and was selected as host to further optimize the production plasmid from nisin-induced expression to constitutive expression with promoters P5 and P8. Constitutive expression can become more favorable than the induced expression as no inducer needs to be added; thus, production costs will be reduced [31]. The results with reporters rfp and lecC showed that with both P5- and P8-derived expression levels, the host L. lactis 9 k-4 can more efficiently produce the reporters than with nisin-induced production. This finding exemplifies that the L. lactis 9 k-4 host with its decreased genome size is a good candidate host for protein production either for cytoplasmic or secreted production with nisin-induced or constitutive expression.
To our knowledge, any unnecessary gene product that is expressed in a production host represents a potential contaminant that can drive up the cost of product purification [32]. Protein purification from spent minimal medium is easy because such media are defined and exhibiting decreased amounts of colored substances that may influence the purification proceses. As such, the genome-streamlined mutant L. lactis 9 k-4 is a promising foundation for further biotechnological applications.
Conclusions
To our knowledge, this study is the first report that determined the contribution of genome reduction on the heterologous protein production in L. lactis. The genome-streamlined L. lactis was constructed as we expected. In addition, some mutants exhibited good properties as hosts for protein production. This finding indicated that the process of introducing further rational deletions based on transcriptome data, gene function information, and comparative genomics to construct simple predictable L. lactis platform for industrial use in future work, according to our specific design, is worthwhile and can contribute to expand the use of L. lactis.
Methods
Bacterial strains, plasmids, and culture conditions
The strains and plasmids used in this work are listed in Table 2. Both L. lactis NZ9000 (L. lactis 9 k) and its derivatives were grown at 30 °C under static condition in M17G media supplemented with 0.5% (w/v) glucose or in defined SA medium supplemented with 0.5% (w/v) glucose [39]. Solid medium contained 1.5% agar. E. coli DH5α was used as intermediate cloning host and was grown aerobically at 37 °C in Luria–Bertani broth (1% tryptone, 0.5% yeast extract, and 1% NaCl). L. lactis 9 k was also used as intermediate cloning host when the plasmid pNZ8048 was used to express the RFP. Listeria monocytogenes WSLC 1019 was cultured aerobically in Brain Heart Infusion (BHI) broth (Sigma, St. Louis, USA) at 37 °C and was used as indicator strain to detect the antibacterial activity of leucocin C. Antibiotic selection was used when appropriate: for E. coli (in milliliters), 150 μg of erythromycin and 15 μg of chloramphenicol; and for L. lactis (in milliliters), 5 μg of erythromycin and 5 μg of chloramphenicol.
DNA manipulations and cloning
DNA markers, T4 DNA ligase, restriction enzymes, and DNA gel extraction kit were purchased from Takara (Dalian, China). PCR product purification kit, First-strand cDNA synthesis kit, and SYBR Green RT-qPCR kit were purchased from Thermo Fisher Scientific (Thermo Fisher Scientific, Waltham, USA). The commercial nisin was purchased from Sigma (St. Louis, USA). L. lactis plasmid DNA, chromosomal DNA, and total RNA were isolated using a Qiaprep spin kit (small scale) following the manufacturer’s instructions. PCRs were performed with the Phusion enzyme (Finnzymes, Espoo, Finland). Primers used in this study are listed in the Additional file 1: Table S2. Primers were purchased from BGI (Beijing, China). Recombinant plasmids were introduced into L. lactis by electrotransformation as previously described [40]. Electrotransformation was performed using a Bio-Rad Gene Pulser (Bio-Rad Laboratories, Richmond, USA). E. coli DH5α was transformed through the CaCl2 procedure [41].
Deletion of large nonessential DNA regions
The sequence of L. lactis 9 k genome was searched and checked on the NCBI website (http://www.ncbi.nlm.nih.gov/nuccore/389853198?report=graph). Several hypothetical proteins and prophage-related proteins were found. Among these genes, five prophages and related proteins (large nonessential DNA regions) were identified, and four large nonessential DNA regions were deleted on schedule with the Cre-loxP system by using the method described previously by our group [21]. The gene knockout vectors pNZ5319△L1, pNZ5319△L2, pNZ5319△L3, and pNZ5319△L4 were constructed subsequently. The first nonessential region was deleted in L. lactis 9 k with the plasmid pNZ5319△L1. The derivative was named as L. lactis 9 k-1. The second nonessential region was then deleted in L. lactis 9 k-1 with the plasmid pNZ5319△L2. The derivative was named L. lactis 9 k-2. The deletion step was repeated with pNZ5319△L3 and pNZ5319△L4 plasmids. The resulting mutants were named L. lactis 9 k-3 and L. lactis 9 k-4, respectively. The correct deletions of the four constructed strains were confirmed by PCR and nucleotide sequence analysis.
Evaluation of physiological profiles
Lactococcus lactis 9 k, L. lactis 9 k-1, L. lactis 9 k-2, L. lactis 9 k-3, and L. lactis 9 k-4 were cultured to OD600 of 0.8 in the M17G medium and diluted to an optical density of OD600 of 0.2. Afterward, 20 μl cultures were incubated into 200 μl M17G and SA media in a 96-well plate. The corresponding bacteria were also incubated into the 100 ml of media in shake flasks. The growth profiles were monitored by measuring OD600 for 16 h at 30 °C by using a Bioscreen machine (Lab-systems, Helsinki, Finland) [23]. Ten milliliter suspension from shake flask was centrifuged when the cells were cultured for 6, 10, and 14 h, and the CDW was calculated using the previously described method [11]. The residual glucose in different strains was detected using commercial kits following the manufacturers’ instructions (Thermo Fisher Scientific, Waltham, USA). The ATP concentration was determined through bioluminescence assay with recombinant firefly luciferase and its substrate d-luciferin by using the Molecular Probes’ ATP Determination Kit (Thermo Fisher Scientific, Waltham, USA). One milliliter cultures of different strains were centrifuged at 6000×g for 5 min before the measurement of ATP concentration. Subsequently, 2 ml of 0.015 g/ml trichloroacetic acid (TCA) was added to the cells and vortexed for 3 min to extract ATP from cells. Afterward, 1 ml of supernatant from vortex was collected and diluted to the concentration of TCA lower than 0.001 g/ml with Tris–acetate buffer (PH 7.8). The extrication of ATP can be used for ATP concentration detection [42, 43]. Each sample was analyzed in triplicate.
Evaluation of phenotype
The metabolism of wild and mutant strains was identified with GP2 MicroPlate™ by the phenotype microarray system (Biolog, California, USA). Sample preparation and assays were conducted in accordance with the manufacturer’s instructions. Briefly, the fresh cultured cells of lactococcal strains on the surface of the solid medium were collected by cotton swab and then dissolved into inoculating fluid (0.40% sodium chloride, 0.03% Pluronic F-68, and 0.02% Gellan Gum) (Biolog, California, USA). Cell density of different strains was equalized to maintain the same number of incubated bacteria. One hundred and fifty microliter tested samples were then pipetted into GP2 plate with various substrates. The plates with the sealing were incubated in the OmniLog® instrument (Biolog, California, USA) at 30 °C for 24 h. The machine automatically recorded the test data once per 30 min. The plates were shaken for 15 s before the data were recorded. Up to two strains can be tested and compared per experiment. Data were analyzed using OL-OM software (version 3.0) (Biolog, California, USA).
Construction of plasmids with red fluorescent protein (rfp) reporter and leucocin C (lecC) reporter
The rfp gene was amplified with pET-28a-rfp plasmid as the template by using the primer pair Prfp-F/Prfp-R. The PCR product rfp gene fragment was digested with KpnI and XbaI and cloned into KpnI-XbaI digested pNZ8048, yielding pNZ8048-rfp plasmid.
The plasmid pLEB690-lecC [19] was digested with HindIII and BamHI. The fragment (PnisZ+Usp45-lecC+lecI) was obtained with DNA gel extraction kit and was cloned into HindIII-BamHI digested pLEB124, yielding pLEB124-lecC (containing the fragment P45+PnisZ+Usp45-lecC+lecI). The strong constitutive promoters P8 and P5 were amplified with the L. lactis N8 chromosomal DNA as the template by using the primer pairs P8-F/R and P5-F/R. P8 and P5 were also cloned into HindIII-BglII digested pLEB124-lecC yielding pLEB124-P8/lecC (containing the fragment P8+PnisZ+Usp45-lecC+lecI) and pLEB124-P5/lecC (containing the fragment P5+PnisZ+Usp45-lecC+lecI), respectively.
The recombinant plasmids pNZ8048-rfp and pLEB124-lecC and control plasmids pNZ8048 and pLEB124 were introduced into L. lactis 9 k, L. lactis 9 k-1, L. lactis 9 k-2, L. lactis 9 k-3, and L. lactis 9 k-4, respectively. The plasmids pLEB124-P8/lecC and pLEB124-P5/lecC were also transformed into L. lactis 9 k-4, respectively. The hosts containing these recombinant plasmids were named as L. lactis 9 k-lecC (or rfp), L. lactis 9 k-1-lecC (or rfp), L. lactis 9 k-2-lecC (or rfp), L. lactis 9 k-3-lecC (or rfp), L. lactis 9 k-4-lecC (or rfp), L. lactis 9 k-4-P8/lecC, and L. lactis 9 k-4-P5/lecC. The commercial nisin was added to the cultures at a concentration of 10 IU/ml to induce lecC expression when the cells were cultured for 5 h [38].
Evaluation of transcriptional efficiency of the rfp and the lecC reporter
The total RNA of different hosts with the same cell density through dilution of mutant strains was extracted from the cells cultured for 6, 10, and 14 h, correspondingly. The transcription of rfp and lecC genes was then analyzed through RT-qPCR with the primer pairs Q-rfp-F/R and Q-lecC-F/R by using the comparative CT (2−△△CT) method with L. lactis tufA gene [44]. L. lactis tufA is a housekeeping gene coding for elongation factor Tu required for the continued translation of mRNA as a control with the primer pair tufA-F/R. All RT-qPCRs were repeated thrice, independently. In addition, a transcription with more than twofold change was regarded as statistically significant [45].
Evaluation of leucocin C antibacterial activity and RFP expression
The antibacterial activity of leucocin C was determined by conventional agar-diffusion method [46]. The indicator strain L. monocytogenes WSLC 1019 was cultured to OD600 of 0.7 in BHI broth. Eight hundred microliters of indicator cultures were added to 100 ml of 60 °C BHI agar media to produce indicator plates. L. lactis 9 k-lecC, L. lactis 9 k-1-lecC, L. lactis 9 k-2-lecC, L. lactis 9 k-3-lecC, L. lactis 9 k-4-lecC, L. lactis 9 k-4-P8/lecC, and L. lactis 9 k-4-P5/lecC were incubated into M17G and SA media. Up to 5 ml cultures of different strains were collected when the cells were cultured for 6, 10, and 14 h. The cell density of mutants was diluted at similar density of the wild strain culture. Supernatants were then collected by centrifugation at 12,000×g for 10 min and were incubated at 70 °C for 15 min. Afterward, 50 μl of supernatants was used in the agar-diffusion experiment. The indicator plates were cultured at 37 °C for 24 h. The expression difference of lecC with nisin to induce expression in different hosts was also analyzed with addition of the commercial nisin to the cultures at a concentration of 10 IU/ml after the cells had been cultured for 5 h. The indicator plates and agar-diffusion experiment were prepared as described above.
L. lactis 9 k-rfp, L. lactis 9 k-1-rfp, L. lactis 9 k-2-rfp, L. lactis 9 k-3-rfp, and L. lactis 9 k-4-rfp were incubated into M17G and SA medium with 96-well plates as described above. The commercial nisin was added to the cultures at a concentration of 2 IU/ml to induce rfp gene expression. The cell density of strains (OD600) and the fluorescence intensity of RFP (excitation: 587 nm; emission: 610 nm) were monitored once every hour by Infinite M200 plate reader (Lab-systems, Helsinki, Finland) [47]. Each sample was analyzed in triplicate. The maximum specific rate (Kmax = △RFP∕△t) of RFP formation in different mutants was calculated during the exponential growth. The difference quotient method was used to calculate the slope of RFP formation. The values of fluorescence intensity at 6, 7, 8 and 9 h were used to calculate the slope of L. lactis 9 k-3-rfp and L. lactis 9 k-4-rfp [Kmax = (△RFP7–6 + △RFP8–7 + △RFP9–8)∕3], respectively, while the values of fluorescence intensity at 8, 9, 10 and 11 h were used to calculate the slope of L. lactis 9 k-rfp, L. lactis 9 k-1-rfp and L. lactis 9 k-2-rfp [Kmax = (△RFP9–8 + △RFP10–9 + △RFP11–10)∕3], respectively.
Statistical analysis
The reported experiments were independently repeated at least three times (as indicated in the text). Data were shown as mean ± standard deviation (SD). All continuous cultivations were carried out in independent biological triplicates, and each sample was additionally taken in technical triplicates. The difference between two groups was compared by t-test defining a P value <0.05 as significant.
Abbreviations
- RFP:
-
red fluorescent protein
- LecC:
-
bacteriocin leucocin C
- CDW:
-
cell dry weight
- Four large nonessential DNA regions:
-
L-1, L-2, L-3, L-4
References
Danchin A. Scaling up synthetic biology: do not forget the chassis. FEBS Lett. 2012;586:2129–37.
Singh V. Recent advancements in synthetic biology: current status and challenges. Gene. 2014;535:1–11.
Levskaya A, Chevalier AA, Tabor JJ, Simpson ZB, Lavery LA, Levy M, Davidson EA, Scouras A, Ellington AD, Marcotte EM, Voigt CA. Synthetic biology: engineering Escherichia coli to see light. Nature. 2005;438:441–2.
Balagadde FK, Song H, Ozaki J, Collins CH, Barnet M, Arnold FH, Quake SR, You L. A synthetic Escherichia coli predator-prey ecosystem. Mol Syst Biol. 2008;4:187.
Shen CR, Lan EI, Dekishima Y, Baez A, Cho KM, Liao JC. Driving forces enable high-titer anaerobic 1-butanol synthesis in Escherichia coli. Appl Environ Microbiol. 2011;77:2905–15.
Moon TS, Yoon SH, Lanza AM, Roy-Mayhew JD, Prather KL. Production of glucaric acid from a synthetic pathway in recombinant Escherichia coli. Appl Environ Microbiol. 2009;75:589–95.
Hashimoto M, Ichimura T, Mizoguchi H, Tanaka K, Fujimitsu K, Keyamura K, Ote T, Yamakawa T, Yamazaki Y, Mori H, et al. Cell size and nucleoid organization of engineered Escherichia coli cells with a reduced genome. Mol Microbiol. 2005;55:137–49.
Petsch D, Anspach FB. Endotoxin removal from protein solutions. J Biotechnol. 2000;76:97–119.
Fischer B, Sumner I, Goodenough P. Isolation, renaturation, and formation of disulfide bonds of eukaryotic proteins expressed in Escherichia coli as inclusion bodies. Biotechnol Bioeng. 1993;41:3–13.
Martinez-Garcia E, Jatsenko T, Kivisaar M, de Lorenzo V. Freeing Pseudomonas putida KT2440 of its proviral load strengthens endurance to environmental stresses. Environ Microbiol. 2015;17:76–90.
Lieder S, Nikel PI, de Lorenzo V, Takors R. Genome reduction boosts heterologous gene expression in Pseudomonas putida. Microb Cell Fact. 2015;14:1.
Morimoto T, Kadoya R, Endo K, Tohata M, Sawada K, Liu S, Ozawa T, Kodama T, Kakeshita H, Kageyama Y, et al. Enhanced recombinant protein productivity by genome reduction in Bacillus subtilis. DNA Res. 2008;15:73–81.
Unthan S, Baumgart M, Radek A, Herbst M, Siebert D, Bruhl N, Bartsch A, Bott M, Wiechert W, Marin K, et al. Chassis organism from Corynebacterium glutamicum—a top-down approach to identify and delete irrelevant gene clusters. Biotechnol J. 2015;10:290–301.
Mierau I, Leij P, van Swam I, Blommestein B, Floris E, Mond J, Smid EJ. Industrial-scale production and purification of a heterologous protein in Lactococcus lactis using the nisin-controlled gene expression system NICE: the case of lysostaphin. Microb Cell Fact. 2005;4:15.
Yin JC, Li GX, Ren XF, Herrler G. Select what you need. A comparative evaluation of the advantages and limitations of frequently used expression systems for foreign genes. J Biotechnol. 2007;127:335–47.
Poquet I, Saint V, Seznec E, Simoes N, Bolotin A, Gruss A. HtrA is the unique surface housekeeping protease in Lactococcus lactis and is required for natural protein processing. Mol Microbiol. 2000;35:1042–51.
Frees D, Savijoki K, Varmanen P, Ingmer H. Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol Microbiol. 2007;63:1285–95.
Morello E, Nouaille S, Cortes-Perez NG, Blugeon S, Medina LF, Azevedo V, Gratadoux JJ, Bermudez-Humaran LG, Le Loir Y, Langella P. Inactivation of the ybdD gene in Lactococcus lactis increases the amounts of exported proteins. Appl Environ Microbiol. 2012;78:7148–51.
Krishnappa L, Monteferrante CG, Neef J, Dreisbach A, van Dijl JM. Degradation of extracytoplasmic catalysts for protein folding in Bacillus subtilis. Appl Environ Microbiol. 2014;80:1463–8.
van Dijl JM, Hecker M. Bacillus subtilis: from soil bacterium to super-secreting cell factory. Microb Cell Fact. 2013;12:1.
Zhu DL, Zhao K, Xu HJ, Zhang XM, Bai YL, Saris PEJ, Qiao MQ. Construction of thyA deficient Lactococcus lactis using the Cre-loxP recombination system. Ann Microbiol. 2015;65:1659–65.
Garcia-Cayuela T, de Cadinanos LP, Mohedano ML, de Palencia PF, Boden D, Wells J, Pelaez C, Lopez P, Requena T. Fluorescent protein vectors for promoter analysis in lactic acid bacteria and Escherichia coli. Appl Microbiol Biotechnol. 2012;96:171–81.
Wan X, Li R, Saris PE, Takala TM. Genetic characterisation and heterologous expression of leucocin C, a class IIa bacteriocin from Leuconostoc carnosum 4010. Appl Microbiol Biotechnol. 2013;97:3509–18.
Zhu DL, Liu FL, Xu HJ, Bai YL, Zhang XM, Saris PEJ, Qiao MQ. Isolation of strong constitutive promoters from Lactococcus lactis subsp lactis N8. Fems Microbiol Letter. 2015;362:fnv107.
Ubeda C, Maiques E, Barry P, Matthews A, Tormo MA, Lasa I, Novick RP, Penades JR. SaPI mutations affecting replication and transfer and enabling autonomous replication in the absence of helper phage. Mol Microbiol. 2008;67:493–503.
Mahony J, Martel B, Tremblay DM, Neve H, Heller KJ, Moineau S, van Sinderen D. Identification of a new P335 subgroup through molecular analysis of lactococcal phages Q33 and BM13. Appl Environ Microbiol. 2013;79:4401–9.
Keravala A, Lee S, Thyagarajan B, Olivares EC, Gabrovsky VE, Woodard LE, Calos MP. Mutational derivatives of PhiC31 integrase with increased efficiency and specificity. Mol Ther. 2009;17:112–20.
Bahey-El-Din M, Griffin BT, Gahan CGM. Nisin inducible production of listeriolysin O in Lactococcus lactis NZ9000. Microb Cell Fact. 2008;7:1.
Hernandez I, Molenaar D, Beekwilder J, Bouwmeester H, Vlieg JETV. Expression of plant flavor genes in Lactococcus lactis. Appl Environ Microbiol. 2007;73:1544–52.
Gao G, Qiao JJ, Yang CH, Jiang DZ, Li RQ, Su JJ, Xu HJ, Zhang XM, Bai YL, Qiao MQ. Functional expression of mouse insulin-like growth factor-I with food-grade vector in Lactococcus lactis NZ9000. Lett Appl Microbiol. 2012;54:404–9.
Berenbaum MR, Zangerl AR. Costs of Inducible Defense: protein limitation, growth, and detoxification in Parsnip Webworms. Ecology. 1994;75:2311–7.
Kolisnychenko V, Plunkett G, Herring CD, Feher T, Posfai J, Blattner FR, Posfai G. Engineering a reduced Escherichia coli genome. Genome Res. 2002;12:640–7.
Woodcock DM, Crowther PJ, Doherty J, Jefferson S, DeCruz E, Noyer-Weidner M, Smith SS, Michael MZ, Graham MW. Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res. 1989;17:3469–78.
Valdesstauber N, Gotz H, Busse M. Antagonistic effect of coryneform bacteria from red smear cheese against Listeria Species. Int J Food Microbiol. 1991;13:119–30.
Qiao M, Immonen T, Koponen O, Saris PE. The cellular location and effect on nisin immunity of the NisI protein from Lactococcus lactis N8 expressed in Escherichia coli and L. lactis. FEMS Microbiol Lett. 1995;131:75–80.
Kuipers OP, de Ruyter PGGA, Kleerebezem M, de Vos WM. Quorum sensing-controlled gene expression in lactic acid bacteria. J Biotechnol. 1998;64:15–21.
Lambert JM, Bongers RS, Kjeerebezem M. Cre-lox-based system for multiple gene deletions and selectable-marker removal in Lactobacillus plantarum. Appl Environ Microbiol. 2007;73:1126–35.
Ra SR, Qiao MQ, Immonen T, Pujana I, Saris PEJ. Genes responsible for nisin synthesis, regulation and immunity form a regulon of two operons and are induced by nisin in Lactococcus lactis N8. Microbiology-Uk. 1996;142:1281–8.
Jensen PR, Hammer K. Minimal requirements for exponential growth of Lactococcus lactis. Appl Microbiol Biotechnol. 1993;59:4363–6.
Holo H, Nes IF. High-frequency transformation, by Electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl Environ Microbiol. 1989;55:3119–23.
Tang X, Nakata Y, Li HO, Zhang M, Gao H, Fujita A, Sakatsume O, Ohta T, Yokoyama K. The optimization of preparations of competent cells for transformation of E. coli. Nucleic Acids Res. 1994;22:2857–8.
Nakamura M, Mie M, Funabashi H, Yamamoto K, Ando J, Kobatake E. Cell-surface-localized ATP detection with immobilized firefly luciferase. Anal Biochem. 2006;352:61–7.
Kimmich GA, Randles J, Brand JS. Assay of picomole amounts of ATP, ADP, and AMP using the luciferase enzyme system. Anal Biochem. 1975;69:187–206.
Castro R, Neves AR, Fonseca LL, Pool WA, Kok J, Kuipers OP, Santos H. Characterization of the individual glucose uptake systems of Lactococcus lactis: mannose-PTS, cellobiose-PTS and the novel GlcU permease. Mol Microbiol. 2009;71:795–806.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–8.
Pongtharangkul T, Demirci A. Evaluation of agar diffusion bioassay for nisin quantification. Appl Microbiol Biotechnol. 2004;65:268–72.
Ungvari Z, Orosz Z, Rivera A, Labinskyy N, Zhao XM, Olson S, Podlutsky A, Csiszar A. Resveratrol increases vascular oxidative stress resistance. AM J Physiol-Heart C. 2007;292:H2417–24.
Authors’ contributions
DLZ carried out experiments, analyzed the primary data and drafted the manuscript. YXF and FLL participated in the construction of the plasmids and gene deletion. HJX, PEJS and MQQ supervised the whole research work and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
None.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its additional files].
Funding
This work was supported by the international cooperation project of MOST (2015DFG32140), the National Natural Science Foundation of China (31470004) and the Academy of Finland Project Number 1268922. Duolong Zhu is supported by the Chinese Scholarship Council (CSC).
Author information
Authors and Affiliations
Corresponding author
Additional file
12934_2016_616_MOESM1_ESM.docx
Additional file 1: Figure S1. Verification of deletions with testing primer pairs. Table S1. Gene content of deleted DNA regions. Table S2. Primers utilized in this study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhu, D., Fu, Y., Liu, F. et al. Enhanced heterologous protein productivity by genome reduction in Lactococcus lactis NZ9000. Microb Cell Fact 16, 1 (2017). https://doi.org/10.1186/s12934-016-0616-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-016-0616-2