
Abstract
Background
Streptomycetes attract a lot of attention in metabolic engineering and synthetic biology because of their well-known ability to produce secondary metabolites. However, the available constitutive promoters are rather limited in this genus.
Results
In this work, constitutive promoters were selected from a pool of promoters whose downstream genes maintained constant expression profiles in various conditions. A total of 941 qualified genes were selected based on systematic analysis of five sets of time-series transcriptome microarray data of Streptomyces coelicolor M145 cultivated under different conditions. Then, 166 putative constitutive promoters were selected by following a rational selection workflow containing disturbance analysis, function analysis, genetic loci analysis, and transcript abundance analysis. Further, eight promoters with different strengths were chosen and subjected to experimental validation by green fluorescent protein reporter and real-time reverse-transcription quantitative polymerase chain reaction in S. coelicolor, Streptomyces venezuelae and Streptomyces albus. The eight promoters drove the stable expression of downstream genes in different conditions, implying that the 166 promoters that we identified might be constitutive under the genus Streptomyces. Four promoters were used in a plug-and-play platform to control the expression of the cryptic cluster of jadomycin B in S. venezuelae ISP5230 and resulted in different levels of the production of jadomycin B that corresponded to promoter strength.
Conclusions
This work identified and evaluated a set of constitutive promoters with different strengths in streptomycetes, and it enriched the presently available promoter toolkit in this genus. These promoters should be valuable in current platforms of metabolic engineering and synthetic biology for the activation of cryptic biosynthetic clusters and the optimization of pathways for the biosynthesis of important natural products in Streptomyces species.
Similar content being viewed by others


Background
Streptomycetes are renowned as a rich source of bioactive natural products of clinical, agricultural, and biotechnological value [1, 2], and they attract large amounts of attention in metabolic engineering and synthetic biology [3]. Promoters play an important role in the aforementioned fields because the fundamental level of transcriptional control takes place at promoter elements that drive gene expression [4, 5]. Therefore, it is crucial to have a powerful promoter toolkit for different purposes of research [6–9].
In streptomycetes, only a limited number of promoters have been described and shown to be functional for the expression of heterologous genes. For example, ermEp*, the constitutive promoters of erythromycin resistance gene (ermE) of Streptomyces erythraeus where the asterisk signifies the presence of a one-base-pair mutation [10]; SF14p, another constitutive promoter isolated form Streptomyces ghanaensis phage I19 [11]; kasOp*, a strong promoter engineered by removing the binding sites of ScbR and ScbR2 from promoter of kasO (sco6280) in Streptomyces coelicolor [12]; tipAp, a thiostrepton-inducible promoter of gene tipA from Streptomyces lividans 66 [13]; and nitAp, an ε-caprolactam-inducible promoter of nitrilase gene (nitA) in Rhodococcus rhodochrous J1 [14]. Among them, ermE*p is the only acknowledged constitutive promoter. Recently, 10 strong promoters in Streptomyces albus were identified in Zhao’s lab, but not all of them were constitutive [15]. Therefore, reliable constitutive promoters are rather limited in streptomycetes.
Constitutive promoters are useful in metabolic engineering and synthetic biology. For instance, constitutive promoters with different strengths are useful for fine-tuning gene expression levels, which can facilitate the pathway optimization of the desired chemicals for higher production [16, 17]. Strong constitutive promoters also can be used to trigger expression of some cryptic clusters, resulting in the discovery of novel natural products [3, 18]. Hence, it is necessary to identify more constitutive promoters in streptomycetes.
Constitutive promoters are usually obtained from the promoters of essential genes whose transcript levels are presumed to be constant [16, 19]. However, for streptomycetes, essential genes are not always constant during the whole life cycle. For example, HrdB (encoded by hrdB) is the essential principle sigma factor and is thought to have stable expression in streptomycetes. However, the transcription of hrdB is regulated by several factors and its transcript level is not always constant [20–22]. Because Streptomyces species have long and complex life cycles, including sophisticated metabolism shifts and morphological differentiation [1], the expression levels of essential genes might be appropriately modulated to adapt to physiological changes. Hence, the traditional approaches to identify constitutive promoters in microbes with short growth periods, such as Escherichia coli and Saccharomyces cerevisiae, may be not reliable for organisms with complex developmental life cycles like streptomycetes.
With the increasing interest in Streptomyces in basic and industrial research, increased transcriptome data are freely available from public databases, especially data from the model organism Streptomyces coelicolor, which offers an opportunity to mine reliable constitutive promoters. In this work, based on the systematic analysis of time-series transcriptome data of the model organism S. coelicolor cultivated in different conditions, a panel of constitutive promoters with different strengths was identified. These promoters were experimentally evaluated by green fluorescent protein (GFP) reporter and real-time reverse-transcription quantitative polymerase chain reaction (RT-qPCR) in S. coelicolor M1146, Streptomyces venezuelae WVR2006, and S. albus J1074; these promoters appeared to be constitutive in different Streptomyces species. These constitutive promoters enriched the promoter toolbox of streptomycetes, which should be of great value for metabolic engineering and synthetic biology in this genus.
Results
Selection of genes with constant expression profiles in S. coelicolor
Constitutive promoters offer relatively constant gene expression profiles. The selection of genes with highly constant expression profiles could allow us to identify constitutive promoters. Therefore, we extracted five sets of time-series transcriptome microarray data of S. coelicolor M145 from the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) database: GSE2983, GSE18489, GSE30569, GSE44415, and GSE53562. The five transcriptome datasets were obtained from the growth of M145 in different conditions, such as medium and culture temperature (Additional file 1: Table S1). Global analysis of gene expression profiles in the five datasets showed that there were 2990, 6019, 5375, 4218, and 4145 genes with constant expression profiles in GSE2983, GSE18489, GSE30569, GSE44415, and GSE53562, respectively (Additional file 2: Dataset S1). To obtain genes with stable expression profiles in the five aforementioned conditions, the intersection of the stably expressed genes in the five datasets was determined, and it contained 941 genes (Fig. 1), implying that the promoters of these genes might be constitutive.

Number of genes with constant expression profiles in S. coelicolor M145. The Venn diagram indicates the number of stably expressed genes based on the time-series transcriptome of M145 cultivated in different conditions
Rational selection of putative constitutive promoters in S. coelicolor
To identify credible constitutive promoters, the 941 genes were further subjected to a series of rational analyses to obtain more reliable genes with stable expression profiles (Fig. 2a). First, internal disturbance analysis was performed to examine whether the expression profiles of the 941 genes remained constant in front of different mutations. Here, the ΔglnK and ΔphoP mutants of S. coelicolor M145 and their corresponding time-series transcriptome microarray datasets GSE30570 and GSE31068 (Additional file 1: Table S1) were used. There were 110 and 195 genes, whose expression profiles were strain-specific in the ΔglnK and ΔphoP mutants, respectively, and were removed from our analysis (Additional file 3: Dataset S2). Then, external disturbance analysis was conducted on the remaining 636 genes (Additional file 4: Dataset S3). Our previous work demonstrated that the type II polyketide jadomycin B (5 μM) can act as an external antibiotic signal to modulate the behaviors of S. coelicolor [23]. Thus, the transcriptome data of S. coelicolor M145 and its ΔscbR2 mutant treated with 5 μM jadomycin B (GSE53563) was used to eliminate genes whose expression levels were sensitive to external stress. In this step, 179 and 76 genes were discarded in M145 and its ΔscbR2 mutant, respectively (Additional file 3: Dataset S2), and the number of the remaining genes was 381 (Additional file 4: Dataset S3).

Rational selection of constitutive promoters in S. coelicolor. a Flow chart of the selection of constitutive promoters. b Transcript levels of genes with stable expression profiles after primary identification (941, black circles) and rational selection (166, blue dots). The red dots indicated genes whose promoters were selected for experiment validation. c The relative strengths of the 166 putative constitutive promoters based on RNA-Seq. The relative strengths of these promoters were reflected by the reads per kilobase per million mapped reads (RPKM) values of their downstream genes. The strength of the promoter of sco0119, which had the lowest RPKM value, was set to one. The red column indicates the experimentally tested constitutive promoters
Functional analysis was carried out to remove genes whose expression profiles were potentially not stable, although they were qualified according to our current conditions. Genes were classified according to Clusters of Orthologous Groups of proteins categories [24]. Here, four genes falling into the category of [Q] (secondary metabolite biosynthesis, transport and catabolism) were excluded (Additional file 3: Dataset S2), because most genes related to secondary metabolites were obviously temporally controlled, which might be ascribed to multiple regulators, as well as complex physiological or environmental signals [23, 25–27]. Another interesting group was the regulators, whose expression profiles are usually condition-dependent and influenced by a highly dynamic complex regulatory network of S. coelicolor [28, 29]. In total, 311 genes (Additional file 4: Dataset S3) were kept after removing the 70 genes (Additional file 3: Dataset S2) that were predicted to be regulators.
There are many cases in which genes in the same operon have different expression profiles [30], implying that there was a potential risk to choosing the promoters of operons as the constitutive promoters. Therefore, genetic loci analysis was performed by predicting operons of M145 based on RNA-Seq (see “Methods”) and removing genes involved in these operons to obtain reliable putative constitutive promoters. There were 145 genes in 127 operons (Additional file 3: Dataset S2), and all of them were eliminated, generating 166 genes with stable expression profiles (Additional file 4: Dataset S3).
According to the genome map of S. coelicolor A3(2), the approximate positions of the promoters of the 166 genes could be determined (Additional file 5: Table S2). Here, the 500-bp sequences upstream of the translation start sites (ATG or GTG) of the 166 genes were extracted for further experiments. These regions contained the full-length promoters and the native 5′ untranslated regions (5′ UTRs) of the corresponding mRNAs. To preliminarily speculate the promoter strength, gene transcript levels reflected by reads per kilobase per million mapped reads (RPKM) (see "Methods") were analyzed based on RNA-Seq; this analysis revealed that the 166 genes varied in a large scope, which approximately covered the abundances of 90 % of all genes in M145 (Fig. 2b). Therefore, we ranked the relative strengths of 166 constitutive promoters according to the RPKM values, and the relative strengths also spanned a wide spectrum (Fig. 2c).
Evaluation of the constitutive promoters in S. coelicolor
To examine whether the putative constitutive promoters obtained by systematic analyses were reliable, eight promoters with different levels of strength were selected for experimental tests. The gfp reporter gene without promoter was used as a control, and the same reporter controlled by the widely used constitutive promoter ermE*p was used as a reference. The eight promoters were evaluated by determining the fluorescence intensity of GFP and the relative mRNA level of gfp in S. coelicolor M1146. This strain was chosen because it is unable to produce actinorhodin (Act), prodiginines (Red), calcium-dependent antibiotic (CDA) and the type I yellow coelicolor polyketide (yCPK) [31], which can significantly reduce the background interference to the fluorescence of GFP.
Samples for experiments were harvested at the exponential phase (24 h), transitional phase (48 h), and stationary phase (60 h) (Additional file 6: Figure S1A). For each strain, there was no significant difference among the intensity of GFP fluorescence in different growth stages, meaning all of the tested promoters drove constant gene expression profiles (Fig. 3a), which was consistent with the stable transcript levels of gfp determined by real-time RT-qPCR (Fig. 4a). The orders of promoter strength reflected by the GFP reporter and real-time RT-qPCR were identical to the result generated by RNA-Seq (RPKM value). Moreover, positive linear relationships (R 2 > 0.85) existed among the RPKM value, relative mRNA level of gfp and fluorescence intensity of GFP (Fig. 5a). These observations demonstrate the high consistency of the results of systematic analysis and experiments, and they suggest that the 166 constitutive promoters might be very reliable in S. coelicolor.

Evaluation of the selected promoters using a GFP reporter. a Fluorescence intensities were determined in S. coelicolor M1146 derivatives with different promoters. b Fluorescence intensities were determined in S. venezuelae WVR2006 derivatives with different promoters. c Fluorescence intensities were determined in S. albus J1074 derivatives with different promoters. “Control” indicates strains with a promoterless GFP reporter. The ermE* promoter was tested in different Streptomyces species for comparison. The values are means ± standard deviation (SD) from three independent experiments. Data obtained from different growth stages of each strain were not statistically significant

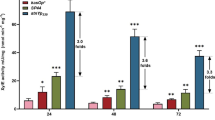
Real-time RT-qPCR assays of the selected promoters. a The relative mRNA of gfp controlled by different promoters in S. coelicolor M1146. b The relative mRNA of gfp controlled by different promoters in S. venezuelae WVR2006. c The relative mRNA of gfp controlled by different promoters in S. albus J1074. The values are means ± SD from three independent experiments. Data obtained from different growth stages of each strain were not statistically significant

Correlations among data related to promoter strengths obtained by GFP reporter, real-time RT-qPCR, and RNA-Seq. a The positive linear relationships of data obtained in S. coelicolor M1146 derivatives. b The positive linear relationships of data obtained in S. venezuelae WVR2006 derivatives. c The positive linear relationships of data obtained in S. albus J1074 derivatives. Black dots in the three-dimensional spaces indicate the overall relationship of promoter strengths reflected by different approaches. The red, green, and blue dots scattered in the three different planes indicate the relationships of promoter strengths measured by any two approaches among the three approaches. For each promoter, the data were the averages of strengths generated by three different approaches. R 2 was the linear regression coefficient
Evaluation of the constitutive promoters in other Streptomyces species
To examine whether the 166 constitutive promoters were applicable in the genus of Streptomyces, the eight selected promoters were further experimentally evaluated in S. venezuelae WVR2006 [32] and S. albus J1074 [33]. The experimental designs in the two species were the same as those in S. coelicolor M1146. Samples were harvested in exponential phase, transitional phase, and stationary phase: 14, 20, and 30 h for S. venezuelae WVR2006 and 36, 48, and 72 h for S. albus J1074 (Additional file 6: Figure S1B and C). As shown in Fig. 3b, all eight promoters yielded constant expression of GFP at different growth stages in S. venezuelae WVR2006, which also agreed with the stable transcript levels of gfp by real-time RT-qPCR (Fig. 4b). The orders of promoter strength ranked by GFP fluorescence intensity and relative mRNA level of gfp were both in accordance with that sequenced with RPKM values (RNA-Seq), and positive linear correlations (R 2 > 0.78) were also observed among the results generated by the three different approaches (Fig. 5b). As we speculated, similar results were also obtained in S. albus J1074 (Figs. 3c, 4c, 5c). The highly consistent results generated in different Streptomyces species indicated that the eight chosen promoters, as well as the rest of the identified promoters, might be reliable constitutive promoters in this genus.
Engineering the production of jadomycin B in S. venezuelae
To demonstrate an application of the above-evaluated constitutive promoters, they were used to control the production of jadomycin B, which is a type II polyketide antibiotic and is synthesized by a cryptic cluster in S. venezuelae ISP5230 [34]. As all 22 structural genes (from jadJ to jadV) in jad cluster are co-transcribed [35], we placed heterologous promoters upstream of jadJ in this work to drive the whole jadomycin biosynthetic cluster (Fig. 6a). Among the eight evaluated constitutive promoters, Psco4503 had the lowest promoter strength, and Psco2468, Psco2074, and Psco5768 that were stronger than ermE*p were selected. Controlled by the strongest constitutive promoter Psco5768, the production of jadomycin B reached 44 mg/L, which was approximately twofold of that controlled by ermE*p. Meanwhile, there was a 5.5-fold range of production generated by the strongest (Psco5768) and weakest (Psco4503) constitutive promoter (Fig. 6b). These results suggested that the 166 constitutive promoters identified in this work might have a greater range of promoter strengths, which might fulfill the requirements for fine-tuning gene expression in metabolic engineering and synthetic biology in the genus Streptomyces.

Engineering of S. venezuelae ISP5230 using constitutive promoters with different strengths to produce jadomycin B. a Diagram for the construction of S. venezuelae ISP5230 derivatives with different constitutive promoters by single-crossover recombination. b Production of jadomycin B driven by different promoters. “Control” indicates that there was no promoter upstream of jadJ. “ND” means that jadomycin B was not detected. The values are means ± SD from three independent experiments
Discussion
Constitutive promoters are crucial in metabolic engineering and synthetic biology. In general, constitutive promoters should maintain constant expression that is independent of internal and external variations, such as life cycle and culture conditions. The lack of available constitutive promoters in streptomycetes urges us to enrich this toolbox for Streptomyces species.
Recently, researchers have mined constitutive promoters from genome-scale data of S. albus, and four of 10 tested promoters were confirmed to be constitutive [15]. The relatively high false-positive rate in the selection of constitutive promoters might be ascribed to the limitations of the transcriptome data, which were obtained from growth in two target culturing conditions and at two defined time points and were inadequate to mimic the profiles of gene transcripts in various conditions. Therefore, we used eight sets of time-series transcriptome microarray data, including five datasets obtained from growth in different media and three datasets obtained from growth in different disturbances, to identify reliable genes with stable expression profiles in various growth stages and culture conditions. Consequently, all of the eight selected promoters from the 166 identified promoters proved to be constitutive in different conditions (Figs. 3, 4, 5), implying that the 166 promoters that we identified are likely to be constitutive in the genus Streptomyces. Therefore, our strategy to identify of constitutive promoters in this work was reliable.
The 500-bp sequences upstream of the translation start site of the 166 genes were cloned for promoter evaluation. This region contained the native 5′ UTR of an mRNA and the full-length promoter [15] to ensure that the putative constitutive promoters functioned well. It was also necessary to use the native 5′ UTR to enable the comparison of promoter strengths in diverse backgrounds because the 5′ UTR plays an important role in maintaining the stability of mRNA in prokaryotes [36, 37]. As suspected, the relative promoter strengths generated by the GFP reporter, real-time RT-qPCR, and RNA-Seq were consistent in different tested Streptomyces species (Fig. 5). These results suggested that the length of the promoters that we selected might ensure the constant expression of the downstream genes in Streptomyces with diverse genetic backgrounds, and the relative strengths of these constitutive promoters were comparable to those in S. coelicolor M145.
Among the eight tested constitutive promoters, only three of them possessed higher strength than ermE*p. One reason for this was that we primarily focused on the selection of reliable constitutive promoters based on genes with highly constant expression profiles, while promoters that possessed a high strength within a narrow time window were filtered by the strict cutoff principles. When necessary, stronger constitutive promoters could also be obtained by further promoter engineering [4]. Actually, the 166 constitutive promoters that we identified possessed a wide spectrum of promoter strengths, which should be a valuable resource to meet various requirements of metabolic engineering and synthetic biology in the genus Streptomyces.
Conclusions
In summary, by taking advantage of transcriptome data of the model organism S. coelicolor, we were able to select regions of the 166 constitutive promoters by following a strict and rational workflow. Eight of the promoters were experimentally evaluated in three different Streptomyces species, and the high consistencies among the data obtained by different approaches implied that the 166 promoters might be reliable constitutive promoters in this genus. Four of the evaluated promoters were used to trigger the cryptic cluster of jadomycin B, and the production of jadomycin B was positively correlated with the promoter strength, suggesting that these constitutive promoters with a wide range of strength might be valuable in metabolic engineering and synthetic biology. Moreover, the systematic and rational strategy employed in this work could be used as a reference for constitutive promoter mining in other organisms.
Methods
Bacterial strains and growth conditions
Plasmids and strains used in this work are listed in Table 1. All plasmids were propagated in E. coli JM109 (Novagen) cultured in Luria–Bertani (LB) broth with 25 μg/mL apramycin at 37 °C. E. coli ET12567/pUZ8002 for conjugation was grown in LB at 37 °C supplemented with antibiotics (chloramphenicol, 25 μg/mL; kanamycin, 25 μg/mL). For spore preparations, S. coelicolor M145 [2], M1146 [31], S. albus J1074 [33] and their derivatives were maintained on mannitol-soya flour (MS) agar plates, while S. venezuelae ISP5230 [38], S. venezuelae WVR2006 [32], and their derivatives were grown on maltose-yeast extract-malt extract (MYM) agar plates. The conjugation of all Streptomyces strains were implemented on MS agar plates with nalidixic acid (25 μg/mL) and apramycin (25 μg/mL). For fermentations, the derivatives of S. coelicolor M1146, S. venezuelae WVR2006 and S. albus J1074 were shaken at 250 rpm in the liquid supplemented minimal medium (SMM), MYM, and tryptic soy broth (TSB), respectively. All cultivations were carried out at 28 °C.
Analysis of microarray transcriptome data of S. coelicolor
All of the transcriptome microarrays were available from the NCBI GEO database with the accession numbers GSE2983, GSE18489, GSE30569, GSE44415, GSE30570, GSE31068, GSE53562, and GSE53563 (Additional file 1: Table S1). For each transcriptome microarray, the transcript signals of each gene were normalized to that obtained at the first sampling time point (or control). If all of the fold changes (increased or decreased) of a gene were no more than two (\( \left| {{ \log }_{2}^{\text{fold change}} } \right| \le 1 \)), this gene was thought to have a constant expression profile, and its promoter was considered to be constitutive.
Evaluation of promoter strength based on RNA-Seq
Cultures of M145 in liquid SMM were sampled at 24 h and used for massively parallel cDNA sequencing. The cDNA libraries were prepared and analyzed on Illumina HiSeq 2000 platform. Samples were sequenced twice to obtain appropriate deep sequencing results. Raw data were processed by removing those with low quality (phred quality <5) and sequencing adaptors. The remaining clean reads were aligned to the edited genome of S. coelicolor. Mapping the total number of reads to each gene was implemented by Picard tools (http://picard.sourceforge.net/). Promoter strength could be reflected by RPKM, which was calculated with the following formula [39].
Operon analysis based on RNA-Seq
Operons were predicted by DOOR 2.0 [40] and Rockhopper [41] based on RNA-Seq data and manually confirmed.
Construction of plasmids and strains
Primers used for plasmid and strain construction are listed in Additional file 7: Table S3. To characterize the identified the promoters, plasmid pIJ8660 [42], which can integrated into the Streptomyces chromosome by site-specific recombination at the phage Ф31 attachment site, was amplified using primers IJF and IJR to generate the linear fragment pIJ8660 containing the gfp reporter gene. By using the genomic DNA of S. coelicolor M145 as template, the 500-bp upstream regions of gene sco4503, sco4508, sco6740, sco3410, sco4658, sco2468, sco2074 and sco5768 were amplified with primer pairs of 4503F and 4503R, 4508F and 4508R, 6740F and 6740R, 3410F and 3410R, 4658F and 4658R, 2468F and 2468R, 2074F and 2074R and 5768F and 5768R, respectively. Promoter ermE* was amplified from pDR4-E using the primer pair ermEF and ermER. Each PCR product contained the promoter and its related 5′ UTR. These PCR products were purified and then linked with pIJ8660 using the Gibson assembly method [43] to generate the target promoters-controlled GFP reporters pIM4503, pIM4508, pIM6740, pIM3410, pIM4658, pIM2468, pIM2074, pIM5768 and pIMermE*, respectively.
To engineer S. venezuelae ISP5230 to enable jadomycin B production using the evaluated promoters, the fragment of pIMermE* was amplified using primers LNF and LNR and self-ligated to remove the elements for site-specific recombination, generating pLN0. The fragment of pLN0 was amplified using primers LN0F and LN0R to remove the gene of gfp, and 5′ end of the jadJ gene was amplified using primers JF and JR. Then the two fragments were assembled using the Gibson assembly method to generate pLNermE*. To replace ermE* with other identified promoters, the fragment of pLN1 was treated with BsaI to remove ermE*, and the promoters of sco6740, sco5768, sco2074 and sco2468 were amplified using primers 6740F and 6740R1, 5768F and 5768R1, 2074F and 2074R1 and 2468F and 2468R1, respectively. Then the fragment of pLN1 and the four promoters were assembled using the Gibson assembly method to generate pLNsco6740p, pLNsco5768p, pLNsco2074p, and pLNsco2468p.
The plasmid-containing E. coli JM109 was co-cultured with S. coelicolor, S. venezuelae, and S. albus, generating the corresponding Streptomyces transformants by conjugation between E. coli and Streptomyces species [44]. The recombinants were confirmed by PCR with the primer gfp-cx-R and the corresponding forward primer of the eight genes.
Measurement of fluorescence intensity of GFP
Cultures (1 mL) were sampled at appropriate times, and the supernatant was removed by centrifugation (4 °C, 10,000×g). Cells were washed with deionized water three times. For S. coelicolor derivatives, the pellets were resuspended in 1 mL binding buffer (20 mM Tris–HCl, 0.5 M NaCl, 10 % (v/v) glycerol and 20 mM imidazole, pH 7.4) and subjected to ultrasonication (power 30 W, pulse 10 s, stop 10 s, for 5 min) to generate cell extract. The quantification of GFP fluorescence intensity was implemented by normalizing the fluorescence intensity to the protein concentration of 200 μL total protein [45, 46]. The concentration of the total protein was assayed by the Bradford method [47]. For derivatives of S. venezuelae and S. albus, the GFP fluorescence intensity was quantified by normalizing the fluorescence intensity of 200 μL deionized water resuspensions to the wet weight of cells in 200 μL cultures [45]. The GFP fluorescence was detected using a microplate reader (BioTek, USA) at the absorption wavelengths of 488 and 529 nm. All experiments were repeated in triplicate. Differences among GFP fluorescence intensities obtained at different growth stages were analyzed by one-way analysis of variance (ANOVA), and p value < 0.05 was considered statistically significant.
Total RNA isolation
Cells of S. coelicolor (24, 48, and 60 h), S. venezuelae (14, 20, and 30 h), and S. albus (36, 48, and 72 h) were quickly harvested by fast filtration, flash frozen in liquid nitrogen, and ground into powder for total RNA extraction using TRNzol according to the manufacturer’s instructions (Tiangen, China). The integrity and quantity of the isolated RNA were checked by denaturing agarose gel electrophoresis and NanoDrop 2000 spectrophotometer (NanoDrop Technologies, USA), respectively.
Measurement of mRNA level of gfp
Strains with the GFP reporter were cultured to determine the promoter strength according to the relative mRNA level of gfp. For real-time RT-qPCR experiments, the first-strand synthesis of cDNA was carried out using 1 μg total RNA with a PrimeScript™ RT Reagent kit with gDNA Eraser (TaKaRa, Japan) following the manufacturer’s instructions. Oligonucleotides used for real-time RT-qPCR are listed in Additional file 7: Table S3. The PCR procedures were as follows: reactions were performed in 72-well plates using an ABI7500 (Applied Biosystems, USA). The PCR mixtures were prepared by following the introduction of SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) and ROX plus (TaKaRa, Japan). Each 20-μl reaction contained 10 μL 2 × PreMix, 0.5 μL of each primer (10 μM) and 2 μL fivefold diluted cDNA. The reaction parameters were as follows: 95 °C for 30 s, followed by 45 two-step amplification cycles consisting of denaturation at 95 °C for 5 s, and annealing at 60 °C for 40 s. The amplification specificity of each assay was confirmed by melting curve analysis carried out at 60–95 °C. Results were collected and analyzed using the supporting 7500 software (v2.0.4). The hrdB gene was used as an internal control [12]. All samples were run in triplicate. Differences among the relative levels of gfp obtained at different growth stages were analyzed by one-way ANOVA, and p-value < 0.05 was considered statistically significant.
Measurement of jadomycin B production
The S. venezuelae strains that were engineered for jadomycin B production were cultured in glucose-MOPS medium at 250 rpm and 28 °C for 48 h. Extraction and high-performance liquid chromatography analysis of jadomycin B were performed as described previously [48].
References
Chater KF. Streptomyces inside-out: a new perspective on the bacteria that provide us with antibiotics. Philos Trans R Soc Lond B Biol Sci. 2006;361:761–8.
Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature. 2002;417:141–7.
KomatsuHwang K-S, Kim HU, Charusanti P, Palsson BØ, Lee SY. Systems biology and biotechnology of Streptomyces species for the production of secondary metabolites. Biotechnol Adv. 2014;32:255–68.
Blazeck J, Alper HS. Promoter engineering: recent advances in controlling transcription at the most fundamental level. Biotechnol J. 2013;8:46–58.
Olano C, Lombó F, Méndez C, Salas JA. Improving production of bioactive secondary metabolites in actinomycetes by metabolic engineering. Metab Eng. 2008;10:281–92.
Siegl T, Tokovenko B, Myronovskyi M, Luzhetskyy A. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab Eng. 2013;19:98–106.
Goldbeck CP, Jensen HM, TerAvest MA, Beedle N, Appling Y, Hepler M, et al. Tuning promoter strengths for improved synthesis and function of electron conduits in Escherichia coli. ACS Synth Biol. 2013;2:150–9.
Curran KA, Crook NC, Karim AS, Gupta A, Wagman AM, Alper HS. Design of synthetic yeast promoters via tuning of nucleosome architecture. Nature Commun. 2014;5:4002. doi:10.1038/ncomms5002.
Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc Natl Acad Sci USA. 2005;102:12678–83.
Bibb MJ, Janssen GR, Ward JM. Cloning and analysis of the promoter region of the erythromycin resistance gene (ermE) of Streptomyces erythraeus. Gene. 1985;38:215–26.
Labes G, Bibb M, Wohlleben W. Isolation and characterization of a strong promoter element from the Streptomyces ghanaensis phage I19 using the gentamicin resistance gene (aacC1) of Tn 1696 as reporter. Microbiol. 1997;143:1503–12.
Wang W, Li X, Wang J, Xiang S, Feng X, Yang K. An engineered strong promoter for streptomycetes. Appl Environ Microbiol. 2013;79:4484–92.
Takano E, White J, Thompson CJ, Bibb MJ. Construction of thiostrepton-inducible, high-copy-number expression vectors for use in Streptomyces spp. Gene. 1995;166:133–7.
Herai S, Hashimoto Y, Higashibata H, Maseda H, Ikeda H, Ōmura S, et al. Hyper-inducible expression system for streptomycetes. Proc Natl Acad Sci USA. 2004;101:14031–5.
Luo Y, Zhang L, Barton KW, Zhao H. Systematic identification of a panel of strong constitutive promoters from Streptomyces albus. ACS Synth Biol. 2015;18(4):1001–10.
Sun J, Shao Z, Zhao H, Nair N, Wen F, Xu JH, et al. Cloning and characterization of a panel of constitutive promoters for applications in pathway engineering in Saccharomyces cerevisiae. Biotechnol Bioeng. 2012;109:2082–92.
Blumhoff M, Steiger MG, Marx H, Mattanovich D, Sauer M. Six novel constitutive promoters for metabolic engineering of Aspergillus niger. Appl Microbiol Biotechnol. 2013;97:259–67.
Strakova E, Zikova A, Vohradsky J. Inference of sigma factor controlled networks by using numerical modeling applied to microarray time series data of the germinating prokaryote. Nucleic Acids Res. 2014;42:748–63.
Partow S, Siewers V, Bjørn S, Nielsen J, Maury J. Characterization of different promoters for designing a new expression vector in Saccharomyces cerevisiae. Yeast. 2010;27:955–64.
Craney A, Hohenauer T, Xu Y, Navani NK, Li Y, Nodwell J. A synthetic luxCDABE gene cluster optimized for expression in high-GC bacteria. Nucleic Acids Res. 2007;35:e46.
Tabib-Salazar A, Liu B, Doughty P, Lewis RA, Ghosh S, Parsy M-L, et al. The actinobacterial transcription factor RbpA binds to the principal sigma subunit of RNA polymerase. Nucleic Acids Res. 2013;41:5679–91.
Otani H, Higo A, Nanamiya H, Horinouchi S, Ohnishi Y. An alternative sigma factor governs the principal sigma factor in Streptomyces griseus. Mol Microbiol. 2013;87:1223–36.
Wang W, Ji J, Li X, Wang J, Li S, Pan G, et al. Angucyclines as signals modulate the behaviors of Streptomyces coelicolor. Proc Natl Acad Sci USA. 2014;111:5688–93.
Tatusov R, Fedorova N, Jackson J, Jacobs A, Kiryutin B, Koonin E, et al. The COG database: an updated version includes eukaryotes. BMC Bioinform. 2003;4:41.
Hesketh A, Sun J, Bibb M. Induction of ppGpp synthesis in Streptomyces coelicolor A3(2) grown under conditions of nutritional sufficiency elicits actII-ORF4 transcription and actinorhodin biosynthesis. Mol Microbiol. 2001;39:136–44.
Bibb MJ. Regulation of secondary metabolism in streptomycetes. Curr Opin Microbiol. 2005;8:208–15.
Horinouchi S. A microbial hormone, A-factor, as a master switch for morphological differentiation and secondary metabolism in Streptomyces griseus. Front Biosci. 2002;7:d2045–57.
Huang J, Lih CJ, Pan KH, Cohen SN. Global analysis of growth phase responsive gene expression and regulation of antibiotic biosynthetic pathways in Streptomyces coelicolor using DNA microarrays. Genes Dev. 2001;15:3183–92.
Castro-Melchor M, Charaniya S, Karypis G, Takano E, Hu W-S. Genome-wide inference of regulatory networks in Streptomyces coelicolor. BMC Genom. 2010;11:578.
Blumenthal T, Gleason KS. Caenorhabditis elegans operons: form and function. Nature Rev Genet. 2003;4:110–8.
Gomez-Escribano JP, Bibb MJ. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microbial Biotechnol. 2011;4:207–15.
Fan K, Pan G, Peng X, Zheng J, Gao W, Wang J, et al. Identification of JadG as the B ring opening oxygenase catalyzing the oxidative C-C bond cleavage reaction in jadomycin biosynthesis. Chem Biol. 2012;19:1381–90.
Chater KF, Wilde LC. Streptomyces albus G mutants defective in the SalGI restriction-modification system. J Gen Microbiol. 1980;116:323–34.
Rix U, Zheng J, Remsing Rix LL, Greenwell L, Yang K, Rohr J. The dynamic structure of jadomycin B and the amino acid incorporation step of its biosynthesis. J Am Chem Soc. 2004;126:4496–7.
Zhang Y, Pan G, Zou Z, Fan K, Yang K, Tan H. JadR*-mediated feed-forward regulation of cofactor supply in jadomycin biosynthesis. Mol Microbiol. 2013;90:884–97.
Richards J, Sundermeier T, Svetlanov A, Karzai AW. Quality control of bacterial mRNA decoding and decay. Biochim Biophys Acta. 2008;1779:574–82.
de Ruiz los Mozos I, Vergara-Irigaray M, Segura V, Villanueva M, Bitarte N, Saramago M, et al. Base pairing interaction between 5′- and 3′-UTRs controls icaR mRNA translation in Staphylococcus aureus. PLoS Genet. 2013;9:e1004001.
Yang K, Han L, Ayer SW, Vining LC. Accumulation of the angucycline antibiotic rabelomycin after disruption of an oxygenase gene in the jadomycin B biosynthetic gene cluster of Streptomyces venezuelae. Microbiol. 1996;142:123–32.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Rev Genet. 2009;10:57–63.
Mao X, Ma Q, Zhou C, Chen X, Zhang H, Yang J, et al. DOOR 2.0: presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res. 2014;42:D654–9.
McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, et al. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 2013;41:e140.
Sun J, Kelemen GH, Fernandez-Abalos JM, Bibb MJ. Green fluorescent protein as a reporter for spatial and temporal gene expression in Streptomyces coelicolor A3(2). Microbiol. 1999;145(Pt 9):2221–7.
Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–5.
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. Norwich: The John Innes Foundation; 2000.
Nothaft H, Dresel D, Willimek A, Mahr K, Niederweis M, Titgemeyer F. The Phosphotransferase system of Streptomyces coelicolor is biased for N-Acetylglucosamine metabolism. J Bacteriol. 2003;185:7019–23.
Butler M, Spearman M, Braasch K. Monitoring cell growth, viability, and apoptosis. In: Pörtner R, editor. Animal cell biotechnology. methods in molecular biology. New York: Humana Press; 2014. p. 169–92.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
Chen Y-H, Wang C-C, Greenwell L, Rix U, Hoffmeister D, Vining LC, et al. Functional analyses of oxygenases in jadomycin biosynthesis and identification of JadH as a bifunctional oxygenase/dehydrase. J Biol Chem. 2005;280:22508–14.
Yang K, Han L, Vining LC. Regulation of jadomycin B production in Streptomyces venezuelae ISP5230: involvement of a repressor gene, jadR2. J Bacteriol. 1995;177:6111–7.
Authors’ contributions
SSL and WSW conceived and designed the experiments. SSL and JYW analyzed the transcriptome data and coordinated the main experiments. SSL, WSW, and KQY wrote the manuscript. XL and SLY carried out fermentations. All authors read and approved the final manuscript.
Acknowledgements
We thank Professor Mervyn Bibb of John Innes Centre for the kind donation of S. coelicolor M1146. This work was supported by the National Natural Science Foundation of China (Grant No. 31300052 and 31400034) and Ministry of Science and Technology of China (Grant No. 2013CB734001).
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Shanshan Li and Junyang Wang contributed equally to this work
Additional files
12934_2015_351_MOESM2_ESM.xlsx
Additional file 2: Dataset S1. Genes with constant transcript levels in five time-series microarray datasets and their intersection.
12934_2015_351_MOESM4_ESM.xlsx
Additional file 4: Dataset S3. Genes with highly stable expression profiles in different conditions generated by step-by-step analysis.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, S., Wang, J., Li, X. et al. Genome-wide identification and evaluation of constitutive promoters in streptomycetes. Microb Cell Fact 14, 172 (2015). https://doi.org/10.1186/s12934-015-0351-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-015-0351-0