
Abstract
Nowadays, obesity is seriously increasing in most of the populations all over the world, and is associated with the development and progression of high-mortality diseases such as type-2 diabetes mellitus (T2DM) and its subsequent cardiovascular pathologies. Recent data suggest that both body fat distribution and adipocyte phenotype, can be more determinant for fatal outcomes in obese patients than increased general adiposity. In particular, visceral adiposity is significantly linked to long term alterations on different cardiac structures, and in developed forms of myocardial diseases such as hypertensive and ischaemic heart diseases, and diabetic cardiomyopathy. Interestingly, this depot may be also related to epicardial fat accumulation through secretion of lipids, adipokines, and pro-inflammatory and oxidative factors from adipocytes. Thus, visceral adiposity and its white single-lipid-like adipocytes, are risk factors for different forms of heart disease and heart failure, mainly in higher degree obese subjects. However, under specific stimuli, some of these adipocytes can transdifferentiate to brown multi-mitochondrial-like adipocytes with anti-inflammatory and anti-apoptotic proprieties. Accordingly, in order to improve potential cardiovascular abnormalities in obese and T2DM patients, several therapeutic strategies have been addressed to modulate the visceral and epicardial fat volume and phenotypes. In addition to lifestyle modifications, specific genetic manipulations in adipose tissue and administration of PPARγ agonists or statins, have improved fat volume and phenotype, and cardiovascular failures. Furthermore, incretin stimulation reduced visceral and epicardial fat thickness whereas increased formation of brown adipocytes, alleviating insulin resistance and associated cardiovascular pathologies.
Similar content being viewed by others


Background
In 2015, the World Health Organization estimated a worldwide population of 2.3 billion of overweight individuals and more than 700 million of obese adults (http://www.who.int/topics/obesity/en/). In high-income countries, the overall rates are more than four times than those detected in lower and lower-middle income countries, though obesity is dangerously growing in Southeast Asia and Latin America. In addition, this dread is not restricted to adulthood, since at least 41 million children are obese or overweight [1]. Obesity has emerged as one of the most critical global health care problems, being largely associated with multiple pathologies such as insulin resistance, type-2 diabetes mellitus (T2DM), metabolic dysfunction, and several cardiovascular injuries (i.e., acute myocardial infarct, diabetic cardiomyopathy, atherothrombosis), which may lead to heart failure (HF) and death. In this sense, a weight loss can be potentially reached by lifestyle modifications, and pharmacological and/or surgical interventions, and this may be linked to improvements in cardiovascular function [2]. Interestingly, the most efficient therapies against the development of cardiometabolic pathologies may target the altered composition and distribution of fat stores. In the present review, we will examine the effects and mechanisms of action of excessive visceral fat storing on HF, especially through its influence on epicardial fat depots. In addition, we will also discuss conventional and prospective interventions in obese and T2DM patients to reduce and distribute visceral and epicardial fat repositories in relation with the associated cardiometabolic risk.
Fat distribution and composition
Obesity, particularly in those patients with higher body mass index (BMI) levels (≥30 kg/m2), is linked to increased cardiovascular mortality compared to normal weight (BMI = 18.8–24.9 kg/m2) [3]. However, the heterogeneity of fat composition [white (WAT), brown (BAT) and beige/brite/brown-like (bAT) adipose tissues] and the distribution of these depots, can be more crucial for the development of cardiometabolic disruptions [4, 5]. In physiological conditions, the presence of WAT and BAT in various stores of adipose tissue suggests a direct transformation of differentiated pre-adipocytes (Myf5− and Myf5+, for WAT and BAT, respectively) into mature cells with different morphological and functional characteristics according with location [6]. In general, WAT accumulates excess of energy as single lipid droplets of triglycerides (TAG) within its adipocytes, which express high levels of leptin and exhibit a few variable number of mitochondria (Table 1). WAT weight generally represents as much as 20% of body weight on normal adults and primarily acts as a storage site for fat, preserving supplementary calories for use in times of scarcity [5]. On the other hand, BAT can store lipids in multiple small vacuole inside its smaller multi mitochondrial brown adipocytes. BAT generates non-shivering thermogenesis and energy dissipation by oxidation of glucose and fatty acids, and activation of the mitochondrial transporter uncoupling protein-1 (UCP1), which deviate electron transfer from ATP synthesis to dissipate protons across the inner mitochondrial membrane, producing heat [7].
WAT is found in gluteofemoral (found in the lower-body parts), subcutaneous (immediately below the dermis), and visceral locations [8] (Table 1). This visceral adipose tissue (VAT) surrounds the inner organs and can be divided in intraperitoneal [omental (for stomach and spleen), mesenteric (for intestine) and epiploic (for colon)], retroperitoneal (surrounding the kidneys), gonadal (adhered to the uterus/ovaries or epididymis/testis), and pericardial or epicardial adipose tissue (around heart). Interestingly, this epicardial adipose tissue (EAT) is correlated to VAT and can play an essential role in cardiac function and homeostasis (see later). By contrast, BAT has relatively large depots in infancy but small volume dispersed throughout WAT stores in adults, where it generally locates in suprarenal, paravertebral and supraclavicular regions, as well as areas near large vessels [9]. Finally, the third adipose repository, bAT, has mixed features of both WAT and BAT. bAT is intermediate in size and number of mitochondria and its beige adipocytes could be originated from multipotent pre-adipocytes located in various WAT depots, or from trans-differentiation of a white adipocyte into a beige (and later brown) adipocyte (i.e., WAT-to-BAT trans-differentiation or “fat browning”) (Table 1). bAT expresses also UCP-1 in humans and is mainly sited in inguinal and neck regions to function for adaptive thermogenesis [10].
Visceral adipose tissue overload and cardiovascular risk
In multiple regression analyses, the traditional cardiovascular risk markers BMI, low-density lipoprotein cholesterol (LDL-C), and family history of T2DM, were long-term predictors of accumulation of VAT and subcutaneous fat volumes, from young towards middle age healthy men [11]. However, excessive VAT was more pathogenic than overloaded subcutaneous fat, since VAT closely linked to cardiometabolic abnormalities [12]. In this sense, VAT accumulation has been correlated with increasing incidence of T2DM, T2DM-associated chronic low-grade inflammation, atherogenic dyslipidemia, and hypertension [13, 14]. VAT was also an independent negative marker of peripheral insulin sensitivity [15], which associated with components of the metabolic syndrome (i.e., hyperglycemia, hypertriglyceridemia, and low HDL-C) [16]. Importantly, several clinical studies have linked VAT deposition with HF. The Multi-Ethnic Study of Atherosclerosis (MESA) reported that VAT independently associated with augmented left ventricular concentricity and hypertrophy [17]. The Health ABC [18] and Cardiovascular Health [19] studies demonstrated a positive relationship between VAT and HF, independently of BMI, waist circumference and the waist-hip ratio, as anthropometric surrogates values for predicting VAT accumulation [20].
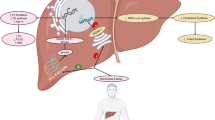
After chronic positive energy balance, WAT adipocytes in VAT lead to free fatty acid (FFA) uptake and accumulation (Fig. 1). WAT expansion in VAT triggered the expression of pro-inflammatory adipokines, oxidative stress and renin-angiotensin-aldosterone system (RAAS) activation. Hypertrophic but not hyperplastic adipocytes, were associated with insulin resistance [21]. Thus, VAT become dysfunctional, dysregulating also adipocyte apoptosis and increasing autophagy [22]. The propensity to preferentially accumulate WAT in VAT stores under conditions of excess energy intake is highly variable from one individual to another. The main etiological factors include age, gender, sex and growth hormones, the endocannabinoid and hypothalamus–pituitary–adrenal systems, glucocorticoids, nutritional factors, and physical activity [23]. Nevertheless, accumulation of fat may saturate VAT capacity. The resultant failure of VAT to store TAG could result in ectopic deposition of toxic fatty acids species (i.e., diacylglycerol, ceramide) in extra-adipose tissue such as myocardium, leading to an increase of EAT thickness [24]. Importantly, the amount of VAT correlates with the volume of EAT, and thus, significant weight loss in obese patients has been associated with noteworthy reduction in the EAT volume [25].

VAT and EAT alterations under obesity and T2DM. In non-obese and non-T2DM subjects, WAT and BAT deposits in both VAT and EAT serve as storages and buffers for fatty acids (FA), attenuators of glycaemia and dyslipidaemia, and as controllers of vascular tension and inflammation. However, under abnormal or excessive fat accumulation, WAT and BAT depots become thicker and dysfunctional. WAT hypertrophies and it saturates, releasing FA and pro-inflammatory factors (cytokines, chemokines, RAAS) towards circulation and myocardium, leading to immune cells (IC) infiltration, myocardial steatosis and insulin resistance. BAT becomes reduced, atrophied and inactive (UCP-1 negative), losing its protective anti-glycemic/dyslipemic and anti-inflammatory effects
Epicardial adipose tissue and cardiac function
EAT is physically next to the myocardium within the lateral wall of the right ventricle and the anterior wall of the left ventricle, surrounding the right coronary artery and the left anterior descending coronary artery [26]. Thus, both EAT and myocardium share the same microcirculation. Computed tomography allows quantification of EAT, which correlates with advancing age and is usually larger in men than in women [25]. EAT displays high rates of WAT lipogenesis but also shows high degrees of WAT lipolysis, serving as local TAG store in metabolic stress and as a buffer for high toxic levels of FFA, in both myocardium and arteries [24] (Fig. 1). EAT may also assist a supportive attenuator of vascular tension, participating in vessel remodelling and paracrine responses by releasing specific molecules for cardiovascular protection. In this sense, adiponectin and adipocyte-derived relaxing factors are discharged to decline contractile and vasoconstrictive effects through endothelium-dependent (i.e., increasing NO/ET-1 ratio) or independent (i.e., reducing cell hyperpolarization and ROS production) mechanisms [27, 28]. Moreover, resident macrophages in EAT can release anti-inflammatory cytokines such as IL-10 [29]. Importantly, EAT transcriptome unveiled that this depot is enriched with genes involved in coagulation, endothelial function, phospholipase activity, apoptosis, and immune signaling [30].
Thus, EAT can protect against myocardial stress, hypertension and local inflammation, and may even function as a BAT store by protecting adjacent tissues from hypothermia because of its small thermogenic adipocytes [31]. In this regard, specific BAT genes such as PR domain containing-16 (PRDM-16), PPAR-γ coactivator 1-α (PGC1α), and UCP-1, are more expressed in EAT than in other fat locations [32]. However, in obesity and T2DM, EAT becomes thicker and dysfunctional, promoting cardiovascular injuries, such as coronary artery disease (CAD) [30, 33].
EAT dysfunction and cardiovascular risk
As VAT, EAT is also subjected to the maladaptive adipocyte biology of obesity, which is characterized by hypertrophy, failure to store TAG, increased lipolysis, and inflammation. A systematic review of several meta-analysis studies showed that EAT was correlated with plasma TAG, fasting glucose, and metabolic syndrome, and it was linked to high systolic blood pressure and CAD [33].
Adipocyte tissue in EAT undergoes FFA uptake, macrophage infiltration, and endothelial cell activation at the heart [34]. A surplus of FFA uptake leads to FFA accumulation through expansion of adipocytes. EAT reached 7.5 mm in thickness in the human metabolic syndrome compared to 4.0 mm in control patients, and this accumulation disturbed insulin resistance in a similar fashion as central fat [35]. Metabolically healthy obese individuals showed more but smaller-sized adipocytes than obese patients with metabolic disorders [36]. Also, rheumatic patients treated with steroids, which are known to imitate some effects of metabolic syndrome, develop thickening of EAT [37]. Thus, threshold values for high-risk echocardiographic EAT measures (over 9.5 and 7.5 mm in men and women, respectively) may be of help for cardiometabolic risk stratification in obese and T2DM subjects [38]. However, EAT quantification is challenging in real clinical practice because of lack of sensitivity and specificity. Imaging acquisition during breath holding and interference of heart beats, water content and fat droplets from parenchymal cells, as well as the biophysical cardiac properties (relaxation times) and field inhomogeneity, can lead to confounding effects and high noise for diagnosis [39]. In this sense, at least in Korean men, increased EAT thickness around the left main coronary artery was not associated with the prevalence of diabetes [40].
Nevertheless, EAT could play an endocrine role over the heart. Dysfunctional adipocytes expressed high levels of pro-inflammatory factors (i.e., IFNγ) that enhanced the pro-inflammatory response of infiltrated immune cells, such as dendritic cells, macrophages, T- and B-cells, and eosinophils [41]. Moreover, accumulated FFA in EAT stimulated macrophages via Toll-like receptor-4 activation, and these macrophages activate pro-inflammatory NF-κB to overexpress chemotactic cytokines (i.e., MCP-1, IL-6) [42]. Consequently, proteomic analysis revealed high levels of anti-oxidant GSTP1, PDIA1, and PGAM1 in EAT compared to subcutaneous adipose tissue in patients with cardiovascular diseases (i.e., cardiomyopathy and CAD), suggesting that EAT suffers greater oxidative stress due to myocardial stress [43]. Then, local inflammation activates resident anti-inflammatory M2 macrophages to pro-inflammatory M1 macrophages, which stimulate cardiac endothelial cells to release more cytokines that reduce insulin signaling in EAT [24]. In this regard, glucose and lipid metabolisms have been shown impaired in EAT of both diabetic and non-diabetic patients with HF [44]. Glucose transport, as well as FFA uptake and re-esterification are decreased, whereas lipolysis is augmented [45]. In addition, anti-inflammatory/-atherogenic adipokines released from EAT (i.e., adiponectin) are also decreased under obesity, contributing to metabolic diseases, and HF [46] (Fig. 1). Particularly, omentin-1, a novel EAT-derived circulating anti-inflammatory and insulin sensitizer adipokine, was reduced in patients with CAD [47].
In addition, EAT can release FFA in the proximity and around the coronaries arteries (perivascular fat), modulating vascular responsiveness to vasoactive agents [48]. EAT may turn into an adverse lipotoxic, pro-thrombotic, and pro-inflammatory organ, being considered a risk factor for CAD and CAD severity [49]. In this regard, EAT-released glycoprotein orosomucoid is an indicator of pro-inflammatory endothelial dysfunction in patients with T2DM or CAD [50]. Also, EAT can discharge FFA into the bloodstream, disturbing vascular homeostasis and endothelial dysfunction, and leading to CAD and hypoxia [51, 52]. In addition, the local RAAS is activated in EAT and contributes to vasoconstriction, inflammation, and following cardiovascular injury [53] (Fig. 1). Finally, due to anatomic proximity of EAT and myocardium and absence of a dividing fascial plane, EAT may also play a key role in myocardial steatosis [24]. The heart possesses an endogenous TAG depot of ≤1.0% organ mass in healthy lean individuals [54]. However, myocardial TAG stores are increased 2 to 4-fold in T2DM and obese patients, which is associated with cardiac hypertrophy and impaired diastolic function [54]. Myocardial steatosis promotes also hypoxia and apoptosis, which strength inflammation [55]. In this line, a bunch of forty-two pro-apoptotic genes (including TNFα and p53) were upregulated in EAT from patients with cardiovascular injuries [56].
Role of BAT on cardiovascular pathophysiology
Remarkably, fat accumulation as BAT may be considered an alternative mean to reduce cardiometabolic risk in obesity and T2DM. Despite its small relative size, BAT is highly vascularized and constitutes an important glucose, fatty acid, and triacylglycerol-clearing organ, and such function could potentially be used to alleviate dyslipidaemias, hyperglycemia, and insulin resistance [57]. Furthermore, BAT influences cardiovascular physiology by releasing factors that regulate vascular tone and both systemic and cardiac metabolisms [9]. BAT stimulation by cold, adrenergic signaling and activators such as thyroid hormones, retinoid, leptin, BMPs, and natriuretic peptides, enhances fatty acid availability for mitochondrial degradation [58, 59] (Fig. 1). Interestingly, BAT lipolysis of stored TAG not only provide an important source of energy but also activate tissue-specific FFA-receptors [53]. Certain adipocyte-specific branch FFA released from BAT diminished adipose tissue inflammation and improved glucose tolerance in obese mice [59, 60]. In this regard, the expression of insulin-sensitive glucose transporter Glut4 has been demonstrated higher in BAT, compared to WAT [59], and specific cytokines discharged from BAT and termed “BATokines”, possess glucose-sensitivity proprieties. For instance, cold-activated BAT secreted fibroblast growth factor-21 (FGF-21) to recover metabolic lipid and glucose equilibrium and leading to cardio-protection in experimental cardiac hypertrophy and ischemia. Also, administration of FGF21 in humans improved hyperlipidemia by lowering plasma TAG and LDL-C levels, while increasing HDL-C levels [61]. Neuregulin-4, a BATokine induced during WAT-to-BAT trans-differentiation, protected against insulin resistance and myocardial ischemia of T2DM mice [62]. Finally, the nerve growth factor (NGF) promoted pro-survival in ischemic cardiomyocytes and diabetic isolated hearts [63].
However, a negative correlation between obesity and levels of BAT volume and activity has been recently stated [64] (Fig. 1). In some South Asians populations, the lower amount of BAT can explain their frequent metabolic and cardiovascular disorders such as obesity, insulin resistance, T2DM, and dyslipidemia [65]. A reduced activity of BAT may predispose subjects to T2DM not only by increasing obesity, but also through a direct pro-diabetic mechanisms, such as by reduction of glucose uptake [59, 64]. BAT in obese/T2DM mice was also less vascularized than in wild type, and their brown adipocytes were larger, unilocular, and mostly UCP1-negative [66]. Thus, conservation of BAT depots with an anti-obesity phenotype may be suggested for therapeutic interventions against cardiovascular pathologies in obese and T2DM patients.
Anti-obesity strategies and reduction of cardiovascular risk
A major goal in the therapeutic field of obesity and related cardiovascular disorders is the development of effective treatments to balance the volume of WAT and BAT, in VAT and EAT stores.
Non-pharmacological reduction of WAT
Changes in nutritional or physical activity are the mainstay intervention for overweight, obese and T2DM patients [25]. In moderate and severe obese patients, a weight loss induced by low-calorie diets and exercise showed reductions of BMI, VAT and EAT (Table 2). Interestingly, EAT shrink in a higher proportion than overall adiposity, and this was significantly associated with cardio-protection [67, 68]. Also, aerobic exercise training significantly increased adiponectin secretion independently of the dietary glycemic index and inversely correlated with VAT shortening [69]. In in vitro assays, a low-calorie diet triggered changes in the secretome of human adipocytes by decreasing secretion of WAT-released pro-inflammatory adipokines [70] (Fig. 2). Furthermore, in selected obese patients with BMI ≥35, a bariatric surgery intervention may be also recommended. Interestingly, after 6–12 months of laparoscopic Roux-en-Y gastric bypass, obese subjects exhibited a substantial decrease in EAT accompanied with VAT, BMI, waist circumference, and cardiovascular risk factors (i.e., total cholesterol, TAG and fasting blood sugar) [71]. Although EAT loss was lower and more limited than VAT, obese patients exhibited higher secretion of adiponectin and leptin, and lessen WAT-related pro-inflammatory adipokines [72, 73]. Intriguingly, the underlying mechanisms of weight loss after bariatric interventions could be more dependent on alteration in gut hormone production [74], neural signalling [75], and glucose/lipid metabolism [76], than those mechanisms related with nutrient absorption.

Prospective fat-modulating interventions for cardiovascular dysfunction in obesity and T2DM. In addition to lifestyle modifications on diet and exercise, cardiovascular complications in patients with increased VAT and EAT may be treated with statins or genetic manipulations (focused on USF1, CIDE-A, PGC1a, UCP-1, PRDM-16 or miR-125b-5p) to decrease WAT thickness/pro-inflammation in EAT or increase WAT browning in VAT, respectively. More significant, PPARγ agonists could promote these effects particularly in EAT, and additionally, incretin stimulation might also induce adipocyte hyperplasia, and subsequent insulin sensitivity
Non-pharmacological stimulation of BAT
Supraclavicular BAT was associated with less obesity and a more favourable metabolic profile in patients with cardiovascular diseases [77], meanwhile severe BAT lipoatrophy aggravated the atherosclerotic process in insulin receptor knockout mice [78]. Consequently, increasing BAT formation and activity may account for novel strategies against obesity and its related cardiometabolic pathologies. In this regard, activation of BAT by β3-adrenergic receptor increased intracellular lipolysis and subsequent replenishment of lipids through de novo lipogenesis and uptake of TAG and cholesterol from circulation. Thus, hyperlipidemic mice were protected from atherosclerosis [79]. Moreover, BAT activation could improve insulin sensitivity via increasing glucose oxidation and lessening body fat mass [80]. Interestingly, high-fat diets stimulated browning capacity of WAT in the retroperitoneal depot by stimulating UCP-1 and CIDE-A (cell death-inducing DFFA-like effector-A) expression, likely, as a compensation mechanism [81]. Also, micro-RNAs such as miR-26, miR-27, mir-30, miR-34a, miR-106b, miR-133, miR-155, miR-193-365, miR-196 and miR-378 have been involved in the control of bAT and BAT formation and function in mice [82].
Therefore, regulation of specific genes or miRs could be used for stimulation of BAT browning in obese and T2DM subjects (Fig. 2). Laurila et al. demonstrated that lacking upstream stimulatory factor 1 (USF1) activated BAT in obese/T2DM mice, and promoted protection against dyslipidaemia, obesity, insulin resistance, and atherosclerosis. These data were also confirmed in subjects carrying a mutation in USF1 [82]. Also, steroid glycosides as ginsenoside Rb1 improved glucose and lipid metabolisms and reduced body weight in obese animals by up-regulating PRDM-16, PGC1α, and UCP-1 expression and WAT browning [83]. Transgenic overexpression of PRDM-16 in subcutaneous WAT protected mice from diet-induced obesity and insulin resistance [84]. In addition, injections of an miR-125b-5p inhibitor directly into WAT increased β3-adrenoceptor-mediated induction of UCP-1 and BAT browning [85].
Furthermore, changes in nutritional or physical activity can also influence on BAT in obese and T2DM individuals (Fig. 2). A potential increase of BAT volume and activity has been postulated with dietary compounds such as vitamin-A and fish oil [86], or exercise training [87]. However, other researchers have demonstrated no change or even decreased BAT activity after exercise [88, 89]. Similarly, the effect of bariatric surgery (i.e., Roux-en-Y gastric bypass or sleeve gastrectomy) in decreasing EAT, is controversial, with a certain variability in the grade of EAT shrinking among the studies [73]. Moreover, activation of BAT has been observed in 40% patients following 1 year of laparoscopic adjustable gastric banding surgery [90]. In this line, experimental BAT transplantation also headed successful outcomes. This procedure augmented intrinsic expression and activity of thermogenic genes in BAT of obese and T2DM mice, and stimulated adiponectin and fatty acid oxidation genes in their WAT. BAT transplantation additionally improved glucose tolerance and decreased insulin resistance, contributing to reduction of liver steatosis and body weight [91]. However, in humans, the amount of BAT is estimated to be less than 0.1% of body weight, which is five times lower than that of mouse [92], and BAT seems less prone to be activated, at least by cold exposure, in obese than in lean subjects [93].
Therefore, we know that WAT accumulation in VAT and EAT is harmful for obesity and related-cardiovascular diseases, and that reduction of these stores or their browning to BAT by changes in nutritional and physical activity can be advantageous. However, hormone production, neural signalling, nutrient absorption and glucose/lipid metabolism could be exclusive for each patient, and unknown (epi)genetic predisposition to obesity and microbiome, may also individually disturb fat storing [94]. Hence, until research progresses on these influencing factors and personalized medicine improves, specific pharmacological approaches could be used to modulate WAT and BAT activity against obesity.
Pharmacological reduction of the WAT/BAT ratio
The most noteworthy treatment for T2DM, metformin, decreased VAT volume, activated BAT and selectively enhanced clearance of VLDL lipoproteins into BAT in obese mice [95] (Table 2). Metformin markedly lowered body weight, plasma cholesterol and TAG, and increased HDL-C levels in obese subjects [96]. However, weight loss could not be achieved in all populations, and fat liver and markers of inflammation and thrombosis were not alleviated [97]. On the other hand, anti-obesity drugs usually work by suppressing appetite, inhibiting fat absorption, or increasing energy consumption and thermogenesis. Unfortunately, some of them (i.e., dexfenfluramine, fenfluramine, sibutramine) have been withdrawn from market because of cardiotoxic side effects [98] Similarly, a PPARγ activator (pioglitazone) was associated with a reduction of pro-inflammatory genes as IL-1β in EAT from T2DM patients with CAD [29]. Also, rosiglitazone triggered lipid turnover and hypolipidemic actions by rapid browning of WAT in EAT depots of obese/T2DM rats by upregulation of PRDM-16, UCP-1 and mitochondrial biogenesis factors (i.e., PGC-1α, COX-4) [99] (Fig. 2). However, PPARγ activators have ben related with cardio-pathological secondary effects in T2DM patients [100].
Thus, only five anti-obesity drugs have been approved by the FDA for long-term treatments [101], but their role in VAT, and overall EAT, is rather unknown. An inhibitor of pancreatic lipase, orlistat, shrink 24% VAT volume in parallel to total cholesterol, LDL-C, TAG, and fasting blood glucose [102]. An agonist of serotonin receptor, lorcaserin, promoted weight loss in T2DM and non-diabetic mainly from the central region of the body [103]. Combination therapies may increase efficacy through synergistic actions, decreases adverse effects and increases tolerability. Thus, naltrexone + bupropion (opioid antagonist/amphetamine) demonstrated a reduction of body weight and VAT, and improved cardiovascular and metabolic parameters, such as blood pressure, lipids and glycaemia [104]. In overweight and obese/T2DM subjects, phentermine + topiramate (meta-amphetamine/monosaccharide) ameliorated body weight and obesity-associated cardio-metabolic conditions, such as blood pressure, total cholesterol and glycated haemoglobin levels [105]. In this sense, since statins have shown pleiotropic effects including the decrease of adipose tissue inflammation, they could also impact the WAT or BAT stores in EAT (Fig. 2). In fact, EAT thickness and inflammation were reduced in T2DM subjects and hyperlipidemic post-menopausal women after atorvastatin administration, independently of lipid lowering or CAD progression [106, 107].
Remarkably, agonists for glucagon-like-protein-1 receptors (GLP-1R) promoted insulin sensitivity, weight loss and adiponectin elevation in obese subjects. They also improved cardiovascular and metabolic parameters, such as blood pressure, lipids and glycaemia [108]. In particular, liraglutide shrink subcutaneous fat [109] and EAT (13%) in T2DM subjects after 12 weeks of treatment [110] (Table 2). In obese/T2DM individuals, liraglutide, but not metformin, reduced 29 and 36% EAT at 3 and 6 months, respectively, after administration [111]. Liraglutide also stimulated WAT browning and thermogenesis in mice independently of nutrient intake [112] (Fig. 2). Another GLP-1R agonist (exenatide), reduced EAT and subcutaneous and liver fat in T2DM patients, in a similar fashion than liraglutide [110]. Also, in obese rodents, exenatide induced a decrease of WAT in VAT and prompted plasma clearance of triacylglycerol and glucose, following BAT activation [112, 113]. In this line, sitagliptin, a DPP-4 inhibitor that avoid GLP-1 degradation, reduced also EAT (15%) in parallel to VAT and more intensively than BMI and waist circumference, in T2DM individuals [114]. Moreover, sitagliptin enhanced energy expenditure in obese mice by UCP-1 up-regulation in BAT repositories [115]. Thus, GLP-1R-associated effects may be also visceral fat specific, and stimulation of incretins could shift the energy balance from obesogenesis to thermogenesis. In this regard, the presence of functional extra-pancreatic GLP-1R has been reported in brain and adipose tissue [116, 117]. GLP-1R at the hypothalamus was crucial for BAT thermogenesis and WAT browning, as well as control of food intake [112, 115]. GLP-1R at the VAT and subcutaneous stores was found elevated in obese and T2DM patients with insulin resistance, where it participated in the overexpression of adiponectin [117, 118]. Finally, GLP-1 also triggered in vitro pre-adipocyte differentiation to stimulate adipocyte hyperplasia and insulin sensitivity [119]. Therefore, incretin may directly target VAT and EAT depots for fat regulation and insulin resistance in obese and T2DM patients.
Conclusions
Adipose tissue may shift from being protective to being detrimental for obesity and cardiovascular homeostasis. WAT in VAT and EAT can hypertrophy and saturate in obese and T2DM subjects, becoming dysfunctional and releasing fatty acid and pro-inflammatory factors, in a positive feed-back loop. In this regard, some additional interventions to life-style change, such as bariatric surgery, BAT transplantation or anti-obesity drugs have exhibited promising outcomes on diminishing the WAT/BAT ratio. Nevertheless, further investigations are needed to discriminate whether this ratio can be specifically amended in EAT. Also, modifications of EAT transcriptome may open new avenues of treatment for cardiometabolic diseases. In these sense, PPARγ agonists and statins could impact on EAT depot by reduction of WAT thickness and pro-inflammation. More significant, incretin stimulation by GLP-1R agonists or DPP-4 inhibitors may reduce the obesogenic phenotype of WAT and encourage its trans-differentiation to BAT, either in VAT and EAT depots, leading to cardiovascular protection (Fig. 2). In addition, DDP4 inhibitors may also contribute to this action by their GLP-1-independent anti-inflammatory properties [120]. Thus, the incretin system may represent a bona fide candidate for improving fat deposition and distribution, and subsequent cardiovascular injuries, in obese and T2DM patients.
References
Bibiloni MDM, Pons A, Tur JA. Prevalence of overweight and obesity in adolescents: a systematic review. ISRN Obes. 2013;2013:392747.
Dallongeville J, Bhatt DL, Steg PHG, Ravaud P, Wilson PW, Eagle KA, et al. Relation between body mass index, waist circumference, and cardiovascular outcomes in 19,579 diabetic patients with established vascular disease: the REACH Registry. Eur J Prev Cardiol. 2012;19:241–9.
Flegal KM, Graubard BI, Williamson DF, Gail MH. Excess deaths associated with underweight, overweight, and obesity. JAMA. 2005;293:1861–7.
Auclair A, Martin J, Bastien M, Bonneville N, Biertho L, Marceau S, Hould FS, Biron S, Lebel S, Lescelleur O, Després JP, Poirier P. Is there a role for visceral adiposity in inducing type 2 diabetes remission in severely obese patients following biliopancreatic diversion with duodenal switch surgery? Obes Surg. 2016;26(8):1717–27.
Cedikova M, Kripnerová M, Dvorakova J, Pitule P, Grundmanova M, Babuska V, et al. Mitochondria in white, brown, and beige adipocytes. Stem Cells Int. 2016;2016:6067349.
Peirce V, Carobbio S, Vidal-Puig A. The different shades of fat. Nature. 2014;510:76–83.
Lee Y, Willers C, Kunji ERS, Crichton PG. Uncoupling protein 1 binds one nucleotide per monomer and is stabilized by tightly bound cardiolipin. Proc Natl Acad Sci USA. 2015;112:6973–8.
Bjørndal B, Burri L, Staalesen V, Skorve J, Berge RK. Different adipose depots: their role in the development of metabolic syndrome and mitochondrial response to hypolipidemic agents. J Obes. 2011;2011:490650.
Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359.
Lee M-J, Wu Y, Fried SK. Adipose tissue heterogeneity: implication of depot differences in adipose tissue for obesity complications. Mol Aspects Med. 2013;34:1–11.
Skårn SN, Eggesbø HB, Flaa A, Kjeldsen SE, Rostrup M, Brunborg C, Reims HM, Aksnes TA. Predictors of abdominal adipose tissue compartments: 18-year follow-up of young men with and without family history of diabetes. Eur J Intern Med. 2016;29:26–31.
Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu C-Y, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116:39–48.
Wang Y, Rimm EB, Stampfer MJ, Willett WC, Hu FB. Comparison of abdominal adiposity and overall obesity in predicting risk of type 2 diabetes among men. Am J Clin Nutr. 2005;81:555–63.
Meisinger C, Döring A, Thorand B, Heier M, Löwel H. Body fat distribution and risk of type 2 diabetes in the general population: are there differences between men and women? The MONICA/KORA Augsburg cohort study. Am J Clin Nutr. 2006;84:483–9.
Lalia AZ, Dasari S, Johnson ML, Robinson MM, Konopka AR, Distelmaier K, et al. Predictors of whole-body insulin sensitivity across ages and adiposity in adult humans. J Clin Endocrinol Metab. 2016;101:626–34.
Goldani H, Adami FS, Antunes MT, Rosa LH, Fassina P, Quevedo Grave MT, et al. Applicatility of the visceral adiposity index (VAI) in the prediction of the components of the metabolic syndrome in elderly. Nutr Hosp. 2015;32:1609–15.
Abbasi SA, Hundley WG, Bluemke DA, Jerosch-Herold M, Blankstein R, Petersen SE, et al. Visceral adiposity and left ventricular remodeling: the Multi-Ethnic Study of Atherosclerosis. Nutr Metab Cardiovasc Dis NMCD. 2015;25:667–76.
Nicklas BJ, Cesari M, Penninx BWJH, Kritchevsky SB, Ding J, Newman A, et al. Abdominal obesity is an independent risk factor for chronic heart failure in older people. J Am Geriatr Soc. 2006;54:413–20.
Djoussé L, Bartz TM, Ix JH, Zieman SJ, Delaney JA, Mukamal KJ, et al. Adiposity and incident heart failure in older adults: the cardiovascular health study. Obesity. 2012;20:1936–41.
Henderson DC, Fan X, Sharma B, Copeland PM, Borba CPC, Freudenreich O, et al. Waist circumference is the best anthropometric predictor for insulin resistance in nondiabetic patients with schizophrenia treated with clozapine but not olanzapine. J Psychiatr Pract. 2009;15:251–61.
Kim JI, Huh JY, Sohn JH, Choe SS, Lee YS, Lim CY, et al. Lipid-overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Mol Cell Biol. 2015;35:1686–99.
Jia G, Jia Y, Sowers JR. Contribution of maladaptive adipose tissue expansion to development of cardiovascular disease. Compr Physiol. 2016;7:253–62.
Tchernof A, Després J-P. Pathophysiology of human visceral obesity: an update. Physiol Rev. 2013;93:359–404.
Fitzgibbons TP, Czech MP. Epicardial and perivascular adipose tissues and their influence on cardiovascular disease: basic mechanisms and clinical associations. J Am Heart Assoc. 2014;3:e000582.
Iacobellis G, Singh N, Wharton S, Sharma AM. Substantial changes in epicardial fat thickness after weight loss in severely obese subjects. Obesity. 2008;16:1693–7.
Iacobellis G. Epicardial and pericardial fat: close, but very different. Obesity. 2009;17:625.
Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119:1661–70.
Salazar J, Luzardo E, Mejías JC, Rojas J, Ferreira A, Rivas-Ríos JR, et al. Epicardial fat: physiological, pathological, and therapeutic implications. Cardiol Res Pract. 2016;2016:1291537.
Sacks HS, Fain JN, Cheema P, Bahouth SW, Garrett E, Wolf RY, et al. Inflammatory genes in epicardial fat contiguous with coronary atherosclerosis in the metabolic syndrome and type 2 diabetes: changes associated with pioglitazone. Diabetes Care. 2011;34:730–3.
McAninch EA, Fonseca TL, Poggioli R, Panos AL, Salerno TA, Deng Y, et al. Epicardial adipose tissue has a unique transcriptome modified in severe coronary artery disease. Obesity. 2015;23:1267–78.
Sacks HS, Fain JN, Bahouth SW, Ojha S, Frontini A, Budge H, et al. Adult epicardial fat exhibits beige features. J Clin Endocrinol Metab. 2013;98:E1448–55.
Sacks HS, Fain JN, Holman B, Cheema P, Chary A, Parks F, et al. Uncoupling protein-1 and related messenger ribonucleic acids in human epicardial and other adipose tissues: epicardial fat functioning as brown fat. J Clin Endocrinol Metab. 2009;94:3611–5.
Rabkin SW. The relationship between epicardial fat and indices of obesity and the metabolic syndrome: a systematic review and meta-analysis. Metab Syndr Relat Disord. 2014;12:31–42.
Ghigliotti G, Barisione C, Garibaldi S, Fabbi P, Brunelli C, Spallarossa P, et al. Adipose tissue immune response: novel triggers and consequences for chronic inflammatory conditions. Inflammation. 2014;37:1337–53.
McLaughlin T, Sherman A, Tsao P, Gonzalez O, Yee G, Lamendola C, et al. Enhanced proportion of small adipose cells in insulin-resistant vs insulin-sensitive obese individuals implicates impaired adipogenesis. Diabetologia. 2007;50:1707–15.
Naukkarinen J, Heinonen S, Hakkarainen A, Lundbom J, Vuolteenaho K, Saarinen L, et al. Characterising metabolically healthy obesity in weight-discordant monozygotic twins. Diabetologia. 2014;57:167–76.
Kitterer D, Latus J, Henes J, Birkmeier S, Backes M, Braun N, et al. Impact of long-term steroid therapy on epicardial and pericardial fat deposition: a cardiac MRI study. Cardiovasc Diabetol. 2015;14:130.
Iacobellis G, Willens HJ, Barbaro G, Sharma AM. Threshold values of high-risk echocardiographic epicardial fat thickness. Obesity. 2008;16:887–92.
Liu C-Y, Redheuil A, Ouwerkerk R, Lima JAC, Bluemke DA. Myocardial fat quantification in humans: evaluation by two-point water-fat imaging and localized proton spectroscopy. Magn Reson Med. 2010;63:892–901.
Chun H, Suh E, Byun AR, Park HR, Shim KW. Epicardial fat thickness is associated to type 2 diabetes mellitus in Korean men: a cross-sectional study. Cardiovasc Diabetol. 2015;14:46.
Xiao L, Yang X, Lin Y, Li S, Jiang J, Qian S, et al. Large adipocytes function as antigen-presenting cells to activate CD4+ T cells via upregulating MHCII in obesity. Int J Obes. 2016;40:112–20.
Kwon H, Pessin JE. Adipokines mediate inflammation and insulin resistance. Front Endocrinol. 2013;4:71.
Salgado-Somoza A, Teijeira-Fernández E, Fernández AL, González-Juanatey JR, Eiras S. Proteomic analysis of epicardial and subcutaneous adipose tissue reveals differences in proteins involved in oxidative stress. Am J Physiol Heart Circ Physiol. 2010;299:H202–9.
Burgeiro A, Fuhrmann A, Cherian S, Espinoza D, Jarak I, Carvalho RA, et al. Glucose uptake and lipid metabolism are impaired in epicardial adipose tissue from heart failure patients with or without diabetes. Am J Physiol Endocrinol Metab. 2016;310:E550–64.
Furuhashi M, Fuseya T, Murata M, Hoshina K, Ishimura S, Mita T, et al. Local production of fatty acid-binding protein 4 in epicardial/perivascular fat and macrophages is linked to coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36:825–34.
López-Jaramillo P, Gómez-Arbeláez D, López-López J, López-López C, Martínez-Ortega J, Gómez-Rodríguez A, et al. The role of leptin/adiponectin ratio in metabolic syndrome and diabetes. Horm Mol Biol Clin Investig. 2014;18:37–45.
Du Y, Ji Q, Cai L, Huang F, Lai Y, Liu Y, et al. Association between omentin-1 expression in human epicardial adipose tissue and coronary atherosclerosis. Cardiovasc Diabetol. 2016;15:90.
Henrichot E, Juge-Aubry CE, Pernin A, Pache J-C, Velebit V, Dayer J-M, et al. Production of chemokines by perivascular adipose tissue a role in the pathogenesis of atherosclerosis? Arterioscler Thromb Vasc Biol. 2005;25:2594–9.
Aslanabadi N, Salehi R, Javadrashid A, Tarzamni M, Khodadad B, Enamzadeh E, et al. Epicardial and pericardial fat volume correlate with the severity of coronary artery stenosis. J Cardiovasc Thorac Res. 2014;6:235–9.
Fandiño-Vaquero R, Fernández-Trasancos A, Alvarez E, Ahmad S, Batista-Oliveira AL, Adrio B, et al. Orosomucoid secretion levels by epicardial adipose tissue as possible indicator of endothelial dysfunction in diabetes mellitus or inflammation in coronary artery disease. Atherosclerosis. 2014;235:281–8.
Perticone F, Ceravolo R, Candigliota M, Ventura G, Iacopino S, Sinopoli F, et al. Obesity and body fat distribution induce endothelial dysfunction by oxidative stress: protective effect of vitamin C. Diabetes. 2001;50:159–65.
Steinberg HO, Paradisi G, Hook G, Crowder K, Cronin J, Baron AD. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes. 2000;49:1231–8.
Patel VB, Mori J, McLean BA, Basu R, Das SK, Ramprasath T, et al. ACE2 deficiency worsens epicardial adipose tissue inflammation and cardiac dysfunction in response to diet-induced obesity. Diabetes. 2016;65:85–95.
Szczepaniak LS, Dobbins RL, Metzger GJ, Sartoni-D’Ambrosia G, Arbique D, Vongpatanasin W, et al. Myocardial triglycerides and systolic function in humans: in vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn Reson Med. 2003;49:417–23.
Ramírez E, Klett-Mingo M, Ares-Carrasco S, Picatoste B, Ferrarini A, Rupérez FJ, et al. Eplerenone attenuated cardiac steatosis, apoptosis and diastolic dysfunction in experimental type-II diabetes. Cardiovasc Diabetol. 2013;12:172.
Maghbooli Z, Hossein-Nezhad A. Transcriptome and molecular endocrinology aspects of epicardial adipose tissue in cardiovascular diseases: a systematic review and meta-analysis of observational studies. Biomed Res Int. 2015;2015:926567.
Festuccia WT, Blanchard P-G, Deshaies Y. Control of brown adipose tissue glucose and lipid metabolism by PPARγ. Front Endocrinol. 2011;2:84.
Bordicchia M, Liu D, Amri E-Z, Ailhaud G, Dessì-Fulgheri P, Zhang C, et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest. 2012;122:1022–36.
Orava J, Nuutila P, Lidell ME, Oikonen V, Noponen T, Viljanen T, et al. Different metabolic responses of human brown adipose tissue to activation by cold and insulin. Cell Metab. 2011;14:272–9.
Golozoubova V, Cannon B, Nedergaard J. UCP1 is essential for adaptive adrenergic nonshivering thermogenesis. Am J Physiol Endocrinol Metab. 2006;291:E350–7.
Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, et al. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013;18:333–40.
Liu SQ, Tefft BJ, Roberts DT, Zhang L-Q, Ren Y, Li YC, et al. Cardioprotective proteins upregulated in the liver in response to experimental myocardial ischemia. Am J Physiol Heart Circ Physiol. 2012;303:H1446–58.
Zheng L-R, Zhang Y-Y, Han J, Sun Z-W, Zhou S-X, Zhao W-T, et al. Nerve growth factor rescues diabetic mice heart after ischemia/reperfusion injury via up-regulation of the TRPV1 receptor. J Diabetes Complicat. 2015;29:323–8.
Ishibashi J, Seale P. Beige can be slimming. Science. 2010;328:1113–4.
Boon MR, Bakker LEH, van der Linden RAD, van Ouwerkerk AF, de Goeje PL, Counotte J, et al. High prevalence of cardiovascular disease in South Asians: central role for brown adipose tissue? Crit Rev Clin Lab Sci. 2015;52:150–7.
Bargut TCL, Aguila MB, Mandarim-de-Lacerda CA. Brown adipose tissue: updates in cellular and molecular biology. Tissue Cell. 2016;48:452–60.
Kim M-K, Tanaka K, Kim M-J, Matuso T, Endo T, Tomita T, et al. Comparison of epicardial, abdominal and regional fat compartments in response to weight loss. Nutr Metab Cardiovasc Dis NMCD. 2009;19:760–6.
Kelly KR, Navaneethan SD, Solomon TPJ, Haus JM, Cook M, Barkoukis H, et al. Lifestyle-induced decrease in fat mass improves adiponectin secretion in obese adults. Med Sci Sports Exerc. 2014;46:920–6.
Kim M-K, Tomita T, Kim M-J, Sasai H, Maeda S, Tanaka K. Aerobic exercise training reduces epicardial fat in obese men. J Appl Physiol. 1985;2009(106):5–11.
Renes J, Rosenow A, Roumans N, Noben J-P, Mariman ECM. Calorie restriction-induced changes in the secretome of human adipocytes, comparison with resveratrol-induced secretome effects. Biochim Biophys Acta. 2014;1844:1511–22.
Hernández-Gil DL, Nieves-Rivera JJ, Mora L, Corretjer L, Altieri PI, Suárez A, et al. Metabolic changes after roux-N-Y bariatric surgery in hispanics. Bol Asoc Médica P R. 2015;107:66–9.
Wu F-Z, Huang Y-L, Wu CC, Wang Y-C, Pan H-J, Huang C-K, et al. Differential effects of bariatric surgery versus exercise on excessive visceral fat deposits. Medicine. 2016;95:e2616.
Rabkin SW, Campbell H. Comparison of reducing epicardial fat by exercise, diet or bariatric surgery weight loss strategies: a systematic review and meta-analysis. Obes Rev Off J Int Assoc Study Obes. 2015;16:406–15.
Korner J, Inabnet W, Febres G, Conwell IM, McMahon DJ, Salas R, et al. Prospective study of gut hormone and metabolic changes after adjustable gastric banding and Roux-en-Y gastric bypass. Int J Obes. 2005;2009(33):786–95.
Zagorodnyuk VP, Chen BN, Brookes SJ. Intraganglionic laminar endings are mechano-transduction sites of vagal tension receptors in the guinea-pig stomach. J Physiol. 2001;534:255–68.
Rubino F, Gagner M, Gentileschi P, Kini S, Fukuyama S, Feng J, et al. The early effect of the Roux-en-Y gastric bypass on hormones involved in body weight regulation and glucose metabolism. Ann Surg. 2004;240:236–42.
Franssens BT, Hoogduin H, Leiner T, van der Graaf Y, Visseren FLJ. Relation between brown adipose tissue and measures of obesity and metabolic dysfunction in patients with cardiovascular disease. J Magn Reson Imaging JMRI. 2017. doi:10.1002/jmri.25594.
Gómez-Hernández A, Beneit N, Escribano Ó, Díaz-Castroverde S, García-Gómez G, Fernández S, et al. Severe brown fat lipoatrophy aggravates atherosclerotic process in male mice. Endocrinology. 2016;157:3517–28.
Berbée JFP, Boon MR, Khedoe PPSJ, Bartelt A, Schlein C, Worthmann A, et al. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nat Commun. 2015;6:6356.
Chondronikola M, Volpi E, Børsheim E, Porter C, Annamalai P, Enerbäck S, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes. 2014;63:4089–99.
García-Ruiz E, Reynés B, Díaz-Rúa R, Ceresi E, Oliver P, Palou A. The intake of high-fat diets induces the acquisition of brown adipocyte gene expression features in white adipose tissue. Int J Obes. 2005;2015(39):1619–29.
Laurila P-P, Soronen J, Kooijman S, Forsström S, Boon MR, Surakka I, et al. USF1 deficiency activates brown adipose tissue and improves cardiometabolic health. Sci Transl Med. 2016;8:323ra13.
Mu Q, Fang X, Li X, Zhao D, Mo F, Jiang G, et al. Ginsenoside Rb1 promotes browning through regulation of PPARγ in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2015;466:530–5.
Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest. 2011;121:96–105.
Giroud M, Pisani DF, Karbiener M, Barquissau V, Ghandour RA, Tews D, et al. miR-125b affects mitochondrial biogenesis and impairs brite adipocyte formation and function. Mol Metab. 2016;5:615–25.
Sakamoto T, Takahashi N, Goto T, Kawada T. Dietary factors evoke thermogenesis in adipose tissues. Obes Res Clin Pract. 2014;8:e533–9.
Ignacio DL, Fortunato RS, Neto RA, da Silva Silvestre DH, Nigro M, Frankenfeld TGP, et al. Blunted response of pituitary type 1 and brown adipose tissue type 2 deiodinases to swimming training in ovariectomized rats. Horm Metab Res. 2012;44:797–803.
Wickler SJ, Stern JS, Glick Z, Horwitz BA. Thermogenic capacity and brown fat in rats exercise-trained by running. Metabolism. 1987;36:76–81.
Vosselman MJ, Hoeks J, Brans B, Pallubinsky H, Nascimento EBM, van der Lans AA, et al. Low brown adipose tissue activity in endurance-trained compared with lean sedentary men. Int J Obes. 2005;2015(39):1696–702.
Vijgen GHEJ, Bouvy ND, Teule GJJ, Brans B, Hoeks J, Schrauwen P, et al. Increase in brown adipose tissue activity after weight loss in morbidly obese subjects. J Clin Endocrinol Metab. 2012;97:E1229–33.
Liu X, Wang S, You Y, Meng M, Zheng Z, Dong M, et al. Brown adipose tissue transplantation reverses obesity in Ob/Ob mice. Endocrinology. 2015;156:2461–9.
Rothwell NJ, Stock MJ. Luxuskonsumption, diet-induced thermogenesis and brown fat: the case in favour. Clin Sci Lond Engl. 1979;1983(64):19–23.
El Hadi H, Frascati A, Granzotto M, Silvestrin V, Ferlini E, Vettor R, et al. Infrared thermography for indirect assessment of activation of brown adipose tissue in lean and obese male subjects. Physiol Meas. 2016;37:N118–28.
Remely M, Aumueller E, Jahn D, Hippe B, Brath H, Haslberger AG. Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benef Microbes. 2014;5:33–43.
Geerling JJ, Boon MR, van der Zon GC, van den Berg SAA, van den Hoek AM, Lombès M, et al. Metformin lowers plasma triglycerides by promoting VLDL-triglyceride clearance by brown adipose tissue in mice. Diabetes. 2014;63:880–91.
Quintero-Castillo D, Luz-Araujo H, Guerra-Velázquez M, Reyna-Villasmil E, Santos Bolívar J, Torres-Cepeda D, et al. Lipid profile in obese and non-obese women with polycystic ovary syndrome treated with metformin. Endocrinol Nutr Organo Soc Espanola Endocrinol Nutr. 2010;57:262–7.
Mauras N, DelGiorno C, Hossain J, Bird K, Killen K, Merinbaum D, et al. Metformin use in children with obesity and normal glucose tolerance–effects on cardiovascular markers and intrahepatic fat. J Pediatr Endocrinol Metab JPEM. 2012;25:33–40.
Wooltorton E. Obesity drug sibutramine (Meridia): hypertension and cardiac arrhythmias. Can Med Assoc J. 2002;166:1307–8.
Distel E, Penot G, Cadoudal T, Balguy I, Durant S, Benelli C. Early induction of a brown-like phenotype by rosiglitazone in the epicardial adipose tissue of fatty Zucker rats. Biochimie. 2012;94:1660–7.
Hernandez AV, Usmani A, Rajamanickam A, Moheet A. Thiazolidinediones and risk of heart failure in patients with or at high risk of type 2 diabetes mellitus: a meta-analysis and meta-regression analysis of placebo-controlled randomized clinical trials. Am J Cardiovasc Drugs. 2011;11:115–28.
Onakpoya IJ, Heneghan CJ, Aronson JK. Post-marketing withdrawal of anti-obesity medicinal products because of adverse drug reactions: a systematic review. BMC Med. 2016;14:191.
Dicker D, Herskovitz P, Katz M, Atar E, Bachar GN. Computed tomography study of the effect of orlistat on visceral adipose tissue volume in obese subjects. Isr Med Assoc J. 2010;12:199–202.
Apovian C, Palmer K, Fain R, Perdomo C, Rubino D. Effects of lorcaserin on fat and lean mass loss in obese and overweight patients without and with type 2 diabetes mellitus: the BLOSSOM and BLOOM-DM studies. Diabetes Obes Metab. 2016;18:945–8.
Smith SR, Fujioka K, Gupta AK, Billes SK, Burns C, Kim D, et al. Combination therapy with naltrexone and bupropion for obesity reduces total and visceral adiposity. Diabetes Obes Metab. 2013;15:863–6.
Allison DB, Gadde KM, Garvey WT, Peterson CA, Schwiers ML, Najarian T, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity. 2012;20:330–42.
Park J-H, Park YS, Kim YJ, Lee IS, Kim JH, Lee J-H, et al. Effects of statins on the epicardial fat thickness in patients with coronary artery stenosis underwent percutaneous coronary intervention: comparison of atorvastatin with simvastatin/ezetimibe. J Cardiovasc Ultrasound. 2010;18:121–6.
Alexopoulos N, Melek BH, Arepalli CD, Hartlage G-R, Chen Z, Kim S, et al. Effect of intensive versus moderate lipid-lowering therapy on epicardial adipose tissue in hyperlipidemic post-menopausal women: a substudy of the BELLES trial (Beyond Endorsed Lipid Lowering with EBT Scanning). J Am Coll Cardiol. 2013;61:1956–61.
Hong JY, Park KY, Kim BJ, Hwang WM, Kim DH, Lim DM. Effects of short-term exenatide treatment on regional fat distribution, glycated hemoglobin levels, and aortic pulse wave velocity of obese type 2 Diabetes mellitus patients. Endocrinol Metab. 2016;31:80–5.
Inoue K, Maeda N, Kashine S, Fujishima Y, Kozawa J, Hiuge-Shimizu A, et al. Short-term effects of liraglutide on visceral fat adiposity, appetite, and food preference: a pilot study of obese Japanese patients with type 2 diabetes. Cardiovasc Diabetol. 2011;10:109.
Morano S, Romagnoli E, Filardi T, Nieddu L, Mandosi E, Fallarino M, et al. Short-term effects of glucagon-like peptide 1 (GLP-1) receptor agonists on fat distribution in patients with type 2 diabetes mellitus: an ultrasonography study. Acta Diabetol. 2015;52:727–32.
Iacobellis G, Mohseni M, Bianco SD, Banga PK. Liraglutide causes large and rapid epicardial fat reduction. Obesity. 2017;25:311–6.
Beiroa D, Imbernon M, Gallego R, Senra A, Herranz D, Villarroya F, et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes. 2014;63:3346–58.
López M, Diéguez C, Nogueiras R. Hypothalamic GLP-1: the control of BAT thermogenesis and browning of white fat. Adipocyte. 2015;4:141–5.
Lima-Martínez MM, Paoli M, Rodney M, Balladares N, Contreras M, D’Marco L, Iacobellis G. Effect of sitagliptin on epicardial fat thickness in subjects with type 2 diabetes and obesity: a pilot study. Endocrine. 2016;51(3):448–55.
Shimasaki T, Masaki T, Mitsutomi K, Ueno D, Gotoh K, Chiba S, et al. The dipeptidyl peptidase-4 inhibitor des-fluoro-sitagliptin regulates brown adipose tissue uncoupling protein levels in mice with diet-induced obesity. PLoS ONE. 2013;8:e63626.
Wei Y, Mojsov S. Tissue-specific expression of the human receptor for glucagon-like peptide-I: brain, heart and pancreatic forms have the same deduced amino acid sequences. FEBS Lett. 1995;358:219–24.
Vendrell J, El Bekay R, Peral B, García-Fuentes E, Megia A, Macias-Gonzalez M, et al. Study of the potential association of adipose tissue GLP-1 receptor with obesity and insulin resistance. Endocrinology. 2011;152:4072–9.
Pastel E, Joshi S, Knight B, Liversedge N, Ward R, Kos K. Effects of Exendin-4 on human adipose tissue inflammation and ECM remodelling. Nutr Diabetes. 2016;6:e235.
Yang J, Ren J, Song J, Liu F, Wu C, Wang X, et al. Glucagon-like peptide 1 regulates adipogenesis in 3T3-L1 preadipocytes. Int J Mol Med. 2013;31:1429–35.
Bae EJ. DPP-4 inhibitors in diabetic complications: role of DPP-4 beyond glucose control. Arch Pharm Res. 2016;39:1114–28.
Authors’ contributions
NG and OL designed, discussed and wrote the work. AGB, ZMV and JE provided critical analysis. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by Banco de Santander-Universidad Autónoma (CEAL-AL/2015-17), Esteve Laboratories, and PIE13/00051 and PI14/00386 (IS. Carlos III).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
González, N., Moreno-Villegas, Z., González-Bris, A. et al. Regulation of visceral and epicardial adipose tissue for preventing cardiovascular injuries associated to obesity and diabetes. Cardiovasc Diabetol 16, 44 (2017). https://doi.org/10.1186/s12933-017-0528-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-017-0528-4