
Abstract
Background
People with COPD have been reported to bear a distinct airway microbiota from healthy individuals based on bronchoalveolar lavage (BAL) and sputum samples. Unfortunately, the collection of these samples involves relatively invasive procedures and is resource-demanding, limiting its regular use. Non-invasive samples from the upper airways could constitute an interesting alternative, but its relationship with COPD is still underexplored. We examined the merits of saliva to identify the typical profile of COPD oral bacteria and test its association with the disease.
Methods
Outpatients with COPD and age-sex matched healthy controls were recruited and characterised based on clinical parameters and 16S rRNA profiling of oral bacteria. A clustering analysis based on patients’ oral bacteria beta-diversity and logistic regressions were performed to evaluate the association between oral bacteria composition and COPD.
Results
128 individuals participated (70 patients and 58 controls). Differential abundance analyses showed differences in patients comparable to the ones previously observed in samples from the lower respiratory tract, i.e., an increase in Proteobacteria (particularly Haemophilus) and loss of microbiota diversity. An unsupervised clustering analysis separated patients in two groups based on microbiota composition differing significantly in the frequency of patients hospitalized due to severe acute exacerbation of COPD (AECOPD) and in the frequency of GOLD D patients. Furthermore, a low frequency of Prevotella was associated with a significantly higher risk of recent severe AECOPD and of being GOLD D.
Conclusion
Salivary bacteria showed an association with COPD, particularly with severe exacerbations, supporting the use of this non-invasive specimen for future studies of heterogeneous respiratory diseases like COPD.
Similar content being viewed by others


Background
The respiratory physiology of patients with chronic obstructive pulmonary disease (COPD) hamper mucociliary clearance in the airways which leads to an exceptional opportunity for bacterial proliferation [1] and results in the establishment of a resident community [2].
In accordance, patients with COPD have been reported to bear a distinct airway microbiota from healthy individuals based on bronchoalveolar lavage (BAL) and sputum specimens [3], though a “typical” COPD profile is difficult to assign since it continuously modifies with disease progression [4]. Nevertheless, some consensus exists regarding (i) a positive correlation between disease severity and microbiota composition, e.g., more severe patients are enriched in Proteobacteria (particularly Haemophilus) [5,6,7,8] and (ii) a negative correlation between disease severity and microbiota diversity [4, 5, 9].
However, evidence for clinical implications of these changes in COPD is still lacking, need short and long-term validation but is fundamental as these might be a promising biomarker of the disease.
Unfortunately, induced sputum or BAL collection are relatively invasive and resource-demanding procedures to be routinely performed (e.g. weekly), requiring trained health-care professionals and specialized equipment. Bronchoscopy in patients with COPD carries a significantly higher risk of complications such as pneumonia, respiratory failure and desaturation compared with those with normal lung function [10].Induced sputum collection, although semi-invasive, generally safe and well tolerated, may lead, especially in more debilitated patients, to some discomfort in sample collection [11, 12].
An interesting alternative would be the use of non-invasive specimens from upper airways, e.g. saliva, since the microbiota of upper and lower airways is highly correlated, and shows topological continuity, implying oral bacteria as the major colonizers of the lower airways, through microaspiration [13,14,15]. Consequently, both niches present several overlapping bacterial genera, e.g. Prevotella, Veillonella and Streptococcus, yet the microbiota from lower airways is less diverse and numerous [13,14,15,16,17]. Saliva’s collection is also friendly enough to be performed frequently (e.g., weekly) even in more debilitated patients.
Here, we have explored, for the first time, the merits of saliva, to identify the typical profile oral bacteria in stable COPD and to test its association with the disease. We have started by describing differences between groups of healthy and diseased individuals. Next, we queried the association between oral bacteria and COPD, by performing an unsupervised clustering analysis that allowed the stratification of people with COPD according to oral bacteria composition.
Methods
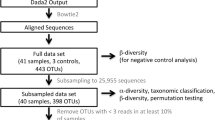
A cross-sectional study was conducted. Ethical approvals were obtained from Administração Regional de Saúde Centro (64/2016) and from Centro Hospitalar do Baixo Vouga (08-03-17). Written informed consent was obtained from all participants. All steps of data collection, processing and analysis were summarized in the Additional file 2.
Subjects and sample collection
Participants with COPD and healthy (controls) were identified by physicians at primary health care centres, hospitals, or senior universities. Patients were eligible if (i) diagnosed with COPD according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria [18], (ii) presented a stable state, with no acute exacerbations in the month prior to enrolment and (iii) were able to give informed consent. Exclusion criteria were (i) presence of severe cardiac, musculoskeletal, or neuromuscular diseases, (ii) cognitive impairment or (iii) active neoplasia or immune diseases. Healthy-individuals were age- and sex-matched to patients with COPD and had similar inclusion and exclusion criteria except for the absence of any respiratory disease. Sociodemographic, anthropometric and clinical data and saliva samples (passive drool) were collected with a structured protocol adapted from the team published work [19]. See Additional file 2 for further details upon data collection. GOLD grades were defined according to FEV1 percentage predicted for each individual. GOLD groups were defined combining the number of exacerbations and hospital admissions of each patient in the year before enrolment with their CAT scores.
DNA extraction
DNA extraction from saliva samples was performed with QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions with slight modifications. DNA quality and quantity was assessed in Denovix DS-11 spectrophotometer. See Additional file 2 for further details.
16S rRNA gene amplification and sequencing
V4 hypervariable region of 16S rRNA gene (F515/R806 primer pair) amplification and sequencing was carried out at the Gene Expression Unit from Instituto Gulbenkian de Ciência, according to the implemented protocol, using Illumina Miseq. See Additional file 2 for further details.
Oral bacteria and statistical analyses
Sample characterisation
Descriptive statistics was used to characterize the sample: comparisons between people with COPD and Healthy controls were conducted with unpaired t-test with Welch’s correction, Mann–Whitney U-test and Chi-square test (statistical analyses conducted in GraphPad Prism 8 [20] and R software v3.6.0 [21]). See Additional file 2 for further details.
Analysis of illumina paired-end reads
QIIME2 2020.8 [22, 23] was used to perform oral bacteria analyses. Quality control procedures were performed via q-score base filtering, chimera removing and 16S-denoising with Deblur [24]. Potential bacterial contaminants were identified with DECONTAM package [25, 26] of R [21] with prevalence method and excluded from subsequent analyses. Taxonomy assignment of amplicon sequence variants (ASVs) was performed with q2-feature-classifier plugin [27, 28], through classify-sklearn method with pre-trained Naïve Bayes classifier against 99% identity eHOMD_v15.1 reference database[29]. All subsequent analyses, except the differential abundance, were performed with data upon ASVs. Differential abundance analyses were done with data upon OTUs at taxonomic level 6 (genus).
Diversity analyses
Alpha-diversity metrics and Beta-diversity metrics were estimated using q2-diversity plugin [30] as implemented in QIIME2 [22, 23]. Spatial dissimilarities between bacterial communities of different groups were assessed with Principal Coordinate Analysis (PCoA) and/or biplots on Weighted Unifrac distance matrix. Mann–Whitney U-test and Kruskal–Wallis with Dunn’s correction were employed to compare alpha-diversity among groups (statistical analyses were performed in GraphPad Prism 8 [20] and R stats package [31] of R [21]). Additionally, the effect of disease state (COPD vs healthy), and cluster segregation (cluster 1 vs cluster 2) on alpha diversity indexes was adjusted for pack-years using a Linear Regression Model (R stats package [31] of R [21]). Permutational multivariate analysis of variance (PERMANOVA) [32, 33] adjusted for pack-years (PY) (vegan package [34] of R [21]) was used to quantify the beta-diversity differences in oral bacteria composition of groups.
Differential abundance analysis of OTUs
Analysis of composition of microbiomes (ANCOM) [35, 36] and Linear discriminant effect size (LefSe) analysis [37, 38] were performed to identify differentially abundant operational taxonomic units (OTUs) between groups of samples and/or clusters. These analyses were conducted with the feature table collapsed at genus taxonomic level (L6). LEfSe was performed in the online version [38] with a linear discriminant analysis (LDA) score of 3 for significance. ANCOM was performed in R with ANCOM 2.0 script[36] with taxa-wise multiple correction and a W cut-off of significance of 0.7. See Additional file 2 for further details.
Clustering analysis
A hierarchical clustering analysis [39] of the oral bacteria (neighbour-joining) based on Weighted Unifrac distance was performed as implemented in QIIME2 [22, 23] under a rarefaction of 4000 sequences per sample and 5000 iterations. See Additional file 2 for further details. Mann–Whitney U-test and Chi-square test (R stats package [31] of R [21]), were used to describe differences in clinical features among different clusters.
Binary logistic regression models and ROC analyses
Binary logistic regression models (glm [40] (link = logit) function of R stats package [31] R software [21], adjusted for PY, were performed to further explore the relation between the most relevant ASVs/OTUs and clinical features in the context of the clustering analysis. Details upon models’ quality assessment were further described in supplementary file. Receiver operating characteristic curves (ROC) and respective discriminatory thresholds were estimated to assess the discriminatory ability of each model (pROC package [41] from R software [21]). Finally, the respective area under the curve (AUC) was also calculated for each model. See Additional file 2 for detailed description of the analyses performed.
Results
Cohort characterisation
Seventy people with COPD (60 male, 68 ± 9y, BMI 25.5 ± 3.5, FEV1pp 48 ± 16, GOLD A-12, B-32, C-5, D-21) and fifty-eight sex and age matched healthy individuals (42 male, 67 ± 8y, BMI 27.6 ± 3.8, FEV1pp 103 ± 17) were included in this study. Detailed characteristics of participants are available in Table 1 and Additional file 1: Table S1.
Oral bacteria composition and diversity are different between people with COPD and healthy controls
Principal coordinate analysis of pairwise distances (Weighted Unifrac) between healthy and people with COPD showed significant differences in oral bacteria composition between groups (PERMANOVA adjusted for PY, p = 0.034) and captured 65% of total diversity (top three principal coordinates).
Oral bacteria of healthy individuals was composed of two major phyla, Firmicutes (40.6%) and Bacteroidetes (30.4%) (Fig. 1a). These were followed by Proteobacteria (16.3%), Fusobacteria (6.7%), Actinobacteria (2.5%) and six low abundant phyla (< 3.5%). In terms of genera, Streptococcus (23%), Prevotella (24%) and Haemophilus (11%) were the most abundant. People with COPD showed a similar oral bacteria composition to healthy individuals, however differences in the relative frequencies of Bacteroidetes (26.5%) and Proteobacteria (22.3%) were observed as well as in genera Prevotella (18%) and Haemophilus (15%).

Salivary microbiota composition and diversity is different between people with COPD and healthy controls. A Mean frequency of phyla and genera of bacteria present in people with COPD and healthy controls. B Cladogram summarizing differentially abundant genera between people with COPD and healthy controls, assessed by LEfSe and ANCOM. Differential genera between groups identified only by LEfSe at a significance cut-off of 3 are represented in black, differential genera pointed by ANCOM at 0.7 significance cut-off are represented in underlined red C Alpha diversity, estimated with Faith’s phylogenetic diversity index, is lower in people with COPD than in healthy controls (Mann–Whitney U-test, U = 1275, p = 0.0013). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Differential abundant bacterial groups between people with COPD and healthy individuals were inferred with LEfSe and ANCOM. Both methods showed that healthy individuals were enriched in Treponema (Spirochaetes), Peptococcus (Firmicutes) and Peptostreptococcus (Firmicutes), whereas according to LEfSe, patients were enriched in genera from Proteobacteria and Firmicutes. Specifically, people with severe airflow obstruction showed an enrichment in Haemophilus, while those with moderate airflow obstruction were enriched in Granullicatella and Lachnoanaerobaculum (see Additional file 2: Fig. S1 for the complete list of genera that differ between the groups).
Oral bacteria of people with COPD was significantly less diverse (Phylogenetic diversity—Alpha diversity, i.e., within individual diversity) than that of healthy individuals (Fig. 1c, Mann–Whitney U test, U = 1275, p = 0.0013). Similar differences were observed after adjusting for PY (ANOVA, F-value = 10.89, p = 0.002).
Oral bacteria composition and diversity are poorly associated with clinical features
We next explored the relationship between oral bacteria and patients’ clinical features. Specifically, we queried whether different levels of airflow obstruction (GOLD grades) and severity of previous exacerbations and symptoms (GOLD groups) were associated with significant differences in oral bacteria diversity and composition.
Considering airflow obstruction, moderate patients (GOLD 1 & 2) showed a significantly distinct oral bacteria composition when compared with severe patients (GOLD 3 & 4) (PERMANOVA adjusted for PY, p = 0.002) but no significant differences were observed in alpha-diversity (Mann–Whitney U-test, U = 435, p = 0.12).
PCoA analysis separated A + B from C + D groups based on the severity of previous exacerbations but not A + C from B + D groups based on the severity of symptoms (PERMANOVA adjusted for PY (A + B vs C + D), p = 0.03, PERMANOVA adjusted for PY (A + C vs B + D), p = 0.06). Alpha-diversity was not significantly different among different levels of severity of previous exacerbations or symptoms (Mann–Whitney U test (A + B vs C + D), U = 420, p = 0.21; Mann–Whitney U test (A + C vs B + D), U = 392, p = 0.64).
No significant associations were found between alpha-diversity and pack-years, hospital admissions, long-term oxygen therapy, treatment with inhaled corticosteroids and SpO2 in people with COPD.
Oral bacteria are associated with disease severity in people with COPD
In an effort to understand to what extent oral bacteria is able to stratify COPD we performed a clustering analysis using the salivary microbial composition of patients. This analysis separated 90% of the individuals in two well supported clusters (“Cluster I” bootstrap node support (bns) = 74% and “Cluster II”, bsn = 84%; Fig. 2) which significantly differed in disease severity.

Unsupervised clustering analysis of the microbiota of people with COPD. A Dendrogram representing Neighbour joining clustering of Weighted Unifrac (samples rarefied by 4000 sequences, with 5000 iteractions). Numbers close to the internal nodes represent bootstrap support. Two major clusters containing 90% of people with COPD emerged: Cluster I and Cluster II. The bar chart represents microbiota composition of each patient at phylum level (Orange—Firmicutes; Blue—Bacteroidetes; Red—Proteobacteria; Green—Fusobacteria; Pink—Actinobacteria). Orange, blue, red and green circles represent the dominant phylum of each sample. The heatmap shows patient status according to “hospital admissions” and “Gold group”. Shading from white to black is proportional to severity level, white less severe and black most severe. B PCoA analysis using Emperor of Weighted UniFrac distance matrix Clusters I and II have a significantly distinct microbiota composition (PERMANOVA adjusted for PY, p = 0.001). Grey arrows represent the 3 most relevant ASVs for cluster segregation. One ASV of Prevotella melaninogenica (d0b698c7298bf04110a6d2f220879bfb) is the major contributor for segregation of Cluster II, while one ASV of Haemophilus parainfluenzae (e27680d4009f98f30248d823bc17fb8e) and another for Streptococcus sp. (a5189f77a2cfeab3bc1602ff5c8ac3e9) contribute for segregation of Cluster I. C The microbiota of Cluster I is less diverse than microbiota of Cluster 2 (Mann–Whitney U-test, U = 271, p = 0.008). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Cluster I aggregated all subjects with a history of recent severe exacerbation leading to hospital admission (Chi-square test, Z = 5.01, p = 0.025)) and 71% of the GOLD D (Chi-square test, Z = 1.98, p = 0.048). Two thirds of those under long term oxygen therapy or with heavier smoking history were also allocated to Cluster I. No other clinical parameters showed significant differences between the two clusters (Additional file 2: Table S2).
Oral bacteria composition was significantly different between the two clusters (PERMANOVA adjusted for PY, P = 0.001. Figure 2b.). Cluster I was enriched in patients dominated by Firmicutes or Proteobacteria, whereas cluster II was mainly represented by patients dominated by Bacteroidetes.
Oral bacteria diversity among patients (alpha diversity) was lower in Cluster I than in Cluster II (Fig. 2c. Mann–Whitney U-test, U = 271, p = 0.008). Similar differences were observed after adjusting for PY (ANOVA, F-value = 5.6, p = 0.006).
Regarding differentially abundant bacteria, both LEfSe and ANCOM distinguished Cluster I as particularly enriched in Streptococcus (Firmicutes) and detected Prevotella and Alloprevotella as responsible for the overabundance of Bacteroidetes in Cluster II (see Additional file 2: Fig. S2 for the complete list of OTUs detected by LEfSe). Both methods further detected a significant enrichment of Dialister (Firmicutes) in Cluster II.
Logistic regression analyses were performed to quantify the risk afforded by the prevalence of Firmicutes, Proteobacteria and Bacteroidetes in oral bacteria of people with COPD belonging to the two clusters. Furthermore, since three ASVs belonging to each of these phyla (Prevotella melaninogenica (Bacteroidetes), Haemophilus parainfluenzae (Proteobacteria) and Streptococcus sp. (Firmicutes)) were the main responsible for cluster segregation (Fig. 2b), the predictive power of their frequency was also inspected.
The combined frequency of Prevotella (Bacteroidetes) and Proteobacteria was found to be the best predictor of being GOLD D, (AUC = 87%), Additional file 2: Table S3 and Fig. 3a), i.e., patients with lower frequency of Prevotella and higher frequency of Proteobacteria were more likely to be severe.

ROC analyses of GOLD D status prediction and recent severe exacerbation status prediction based on logistic regression models adjusted for Pack-years. A ROC curves of GOLD D status prediction based on relative frequency of Prevotella, Proteobacteria and Prevotella + Proteobacteria. The blue curve represents the prediction based on Prevotella relative frequency (AUC = 81%), the red curve represents the prediction based on Proteobacteria relative frequency (AUC = 75%) and the green curve represents the prediction based on the two regressors model (AUC = 87%). B ROC curves of recent severe exacerbation prediction based on relative frequency of Prevotella genus and Prevotella melaninogenica ASV (d0b698c7298bf04110a6d2f220879bfb). Light blue curve represents the prediction made with Prevotella genus relative frequency (AUC = 89%), dark blue curve represents the prediction made with Prevotella melaninogenica ASV relative frequency (AUC = 86%)
Moreover, the odds ratio (OR) of 0.44 suggests a protective effect for increasing frequencies of Prevotella, while the OR of 2.83 suggests a risk effect for increasing frequencies of Proteobacteria.
Prevotella was the best predictor of recent severe exacerbation (leading to hospital admission) (AUC = 89%), which translated into a significantly higher risk for patients with low frequencies of this genus. The OR of 0.58, corroborated the protective effect (Additional file 2: Table S3 and Fig. 3b). Additionally, the frequency of Prevotella melaninogenica alone was also a good predictor for recent severe exacerbation (AUC = 86%) (Additional file 2: Table S3 and Fig. 3b), similarly the OR of 0.63 suggests a protective effect for higher frequencies of this ASV.
No significant associations were found considering Bacteroidetes, Haemophilus, Haemophilus parainfluenzae, Firmicutes, Streptococcus or Streptococcus sp. relative frequencies.
Discussion
Overall, our data suggests that the presence of an abundant Bacteroidetes community (dominated by commensal Prevotella species) in patients’ oral bacteria could have a protective effect towards severe COPD exacerbations.
Strikingly, we observed the separation of patients with recent history of severe exacerbation from all others based on oral bacteria, with low frequencies of Prevotella being the signature of this event. Possible mechanisms underlying this effect include the Prevotella-induced reduction of lung epithelial cell permeability (by modulating the expression of tight junction proteins) [42] or Prevotella-induced microbiota stabilization and resistance to pathobionts colonization [43].
The same phyla dominating the profile of patients with a history of recent severe exacerbation have been previously reported to be enriched in patients with higher predisposition for exacerbations [44, 45].
The depletion of Prevotella and the increased frequencies of Haemophilus and Streptococcus in patients with history of recent severe exacerbations corroborate the vicious cycle hypothesis [46]. According to this theory, the prolonged exposure to tobacco smoke (cluster I aggregated a greater proportion of heavier smokers) induces inflammation in the lung with increased levels of oxidative stress, protease imbalance and mucus hypersecretion. Consequently, an exacerbated, but not efficient, innate immune response allows for facultative anaerobes, e.g. Haemophilus which are better fitted, to persist and proliferate in the lung, enhancing further derangements in innate immunity mechanisms, and possibly triggering COPD exacerbations. Even after smoking cessation, the repetitive cycles of microbiota dysbiosis, together with impaired immune response cause irreversible structural modifications in the small airways and alveoli contributing for COPD progression.
Conversely, less severe patients displayed an overabundance of Prevotella, characteristic of healthy subjects [47].
Prevotella is the most abundant genus in the respiratory tract of healthy individuals [48] with some species having inflammatory properties [49], but most members likely being commensals. Among these, in our study, P. melaninogenica was depleted in the most severe grades of the disease and showed a potential protective effect against severe exacerbations. Interestingly the same species has been previously reported to have a protective role in vitro. By co-cultivating P. melaninogenica and H. influenza, Larsen et al. [50] demonstrated that P. melaninogenica modulated the in vitro inflammatory response of human dendritic cells induced by H. influenzae.
The characterization of oral bacteria of people with COPD and healthy individuals broadly corroborated the main differences previously observed in the lower respiratory tract (e.g. sputum or BAL) [4, 7, 51]: an expansion of Proteobacteria in patients and a Firmicutes and Bacteroidetes enrichment in healthy. Moreover, the overrepresentation of Granullicatella in moderate patients and Haemophilus in severe patients in our study, matches the observations in sputum of people with COPD [4, 7, 51]. Loss of microbial diversity, considered a signature of dysbiosis [52] due to its importance for microbiota stability [53], was also present in people with COPD. This is compatible with less complex airway microbiotas having a lower resistance to colonization by pathobionts [54], such as Haemophilus, frequently implicated in COPD exacerbations.
The microbiota characterization of clinically defined groups lies on the assumption of a correspondence between clinical categories and microbial profiles. Nevertheless, since COPD is a complex disease, enforcing such a correspondence might obscure the relationship between microbiota and the disease.
To explore the potential of the microbiota to stratify the disease, we performed an unsupervised clustering analysis of the diseased population. Interestingly, this analysis separated the individuals in two groups displaying different severities but showed only a week correspondence with the obstruction level, which is the criterion to diagnose the disease.
Some limitations of our study need to be acknowledged. First, although innovative and with great potential as a prognostic biomarker for COPD, the salivary bacterial community is susceptible to be influenced by oral health and smoking habits. For example, periodontitis is likely to influence the salivary microbiota [55] and has been previously associated with COPD [56]. Nevertheless, the bacterial groups found by two recent publications distinguishing the salivary microbiota of healthy individuals from people with periodontitis [55] or from people with periodontitis concomitant with COPD [57], were not coincident with the ones found by our study. Though we cannot discard the influence of this oral disease in our work it does not seem to be a major factor to differentiate patients from healthy.
Second, we were not able to evaluate the prospective effects of high frequencies of Prevotella in terms of preventing severe COPD exacerbations due to the cross-sectional study design. Further studies should explore these effects prospectively.
Third, we acknowledged that the use of saliva is still exploratory and that external validation of our findings in a multicentre trial and with larger cohorts is needed to assess its robustness, especially in a disease as heterogeneous as COPD.
In conclusion, based on the analysis of people with COPD and healthy individuals, our data suggests an association between oral bacteria and COPD, particularly in terms of severe exacerbations. It further shows that even in stability it is possible to identify the dysbiotic microbial signatures associated with severe COPD exacerbations.
Additionally, our results suggest that unsupervised analyses of oral bacteria may provide a more useful insight into its relationship with the disease potentially enabling practical applications such as risk assessment and patient counselling.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article (and its additional file(s)). Furthermore, raw sequencing data is deposited in National Centre for Biotechnology Information's (NCBI) Sequence Read Archive (SRA) (BioProject PRJNA800368).
Change history
21 December 2022
A Correction to this paper has been published: https://doi.org/10.1186/s12931-022-02249-6
References
Rogers GB. The lung microbiome. Emerg Topics Life Sci. 2017;1:313–24.
Dickson RP, Huffnagle GB. The lung microbiome: new principles for respiratory bacteriology in health and disease. PLoS Pathog. 2015;11: e1004923.
Molyneaux PL, Mallia P, Cox MJ, Footitt J, Willis-Owen SAG, Homola D, et al. Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188:1224–31.
Mayhew D, Devos N, Lambert C, Brown JR, Clarke SC, Kim VL, et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax. 2018;73:422–30.
Garcia-Nunez M, Millares L, Pomares X, Ferrari R, Perez-Brocal V, Gallego M, et al. Severity-related changes of bronchial microbiome in chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52:4217–23.
Sze MA, Dimitriu PA, Suzuki M, McDonough JE, Campbell JD, Brothers JF, et al. Host response to the lung microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192:438–45.
Wang Z, Singh R, Miller BE, Tal-Singer R, Van Horn S, Tomsho L, et al. Sputum microbiome temporal variability and dysbiosis in chronic obstructive pulmonary disease exacerbations: an analysis of the COPDMAP study. Thorax. 2018;73:331–8.
Budden KF, Shukla SD, Rehman SF, Bowerman KL, Keely S, Hugenholtz P, et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir Med. 2019;7:907–20.
Galiana A, Aguirre E, Rodriguez JC, Mira A, Santibanez M, Candela I, et al. Sputum microbiota in moderate versus severe patients with COPD. Eur Respir J. 2014;43:1787–90.
Rand IAD, Blaikley J, Booton R, Chaudhuri N, Gupta V, Khalid S, et al. British Thoracic Society guideline for diagnostic flexible bronchoscopy in adults: accredited by NICE. Thorax. 2013;68:i1-44.
Taube C, Holz O, Mücke M, Jörres RA, Magnussen H. Airway response to inhaled hypertonic saline in patients with moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164:1810–5.
Makris D, Tzanakis N, Moschandreas J, Siafakas NM. Dyspnea assessment and adverse events during sputum induction in COPD. BMC Pulm Med. 2006;6:17.
Gaeckle NT, Pragman AA, Pendleton KM, Baldomero AK, Criner GJ. The oral-lung axis: the impact of oral health on lung health. Respir Care. 2020;65:1211–20.
Mammen MJ, Scannapieco FA, Sethi S. Oral-lung microbiome interactions in lung diseases. Periodontol. 2000;2020(83):234–41.
Huffnagle GB, Dickson RP, Lukacs NW. The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol. 2017;10:299–306.
Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–63.
Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio. 2015;6:e00037-e115.
GOLD—Global Strategy for Diagnosis, Management, and prevention of chronic obstructive pulmonary disease 2020 report. 2020. 1–141.
Marques A, Jácome C, Rebelo P, Paixão C, Oliveira A, Cruz J, et al. Improving access to community-based pulmonary rehabilitation: 3R protocol for real-world settings with cost-benefit analysis. BMC Public Health. 2019;19:676.
Prism—GraphPad. https://www.graphpad.com/scientific-software/prism/. Accessed 12 Jan 2022.
R: The R Project for Statistical Computing. https://www.r-project.org/. Accessed 12 Jan 2022.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7.
QIIME 2. https://qiime2.org/. Accessed 12 Jan 2022.
deblur—QIIME 2 2020.8.0 documentation. https://docs.qiime2.org/2020.8/plugins/available/deblur/. Accessed 30 Dec 2021.
Callahan B, Davis NM, Ernst FGM. decontam: Identify contaminants in marker-gene and metagenomics sequencing data. Bioconductor version: Release (3.14); 2021. https://bioconductor.org/packages/decontam/. Accessed 30 Dec 2021.
Callahan B. benjjneb/decontam. 2021. https://github.com/benjjneb/decontam. Accessed 12 Jan 2022.
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6:90.
feature-classifier—QIIME 2 2020.8.0 documentation. https://docs.qiime2.org/2020.8/plugins/available/feature-classifier/?highlight=feature%20classifier. Accessed 12 Jan 2022.
Escapa IF, Huang Y, Chen T, Lin M, Kokaras A, Dewhirst FE, et al. Construction of habitat-specific training sets to achieve species-level assignment in 16S rRNA gene datasets. Microbiome. 2020;8:65.
diversity—QIIME 2 2020.8.0 documentation https://docs.qiime2.org/2020.8/plugins/available/diversity/. Accessed 30 Dec 2021.
R: The R Stats Package. https://stat.ethz.ch/R-manual/R-devel/library/stats/html/00Index.html. Accessed 12 Jan 2022.
Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46.
adonis function—RDocumentation. https://www.rdocumentation.org/packages/vegan/versions/2.4-2/topics/adonis. Accessed 15 Sep 2021.
vegan-package: Community Ecology Package: Ordination, Diversity and... in vegan: Community Ecology Package. https://rdrr.io/cran/vegan/man/vegan-package.html. Accessed 12 Jan 2022.
Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis. 2015;26:27663.
Mandal, Siddhartha. Research—Dr. Siddhartha Mandal: ANCOM 2.0—updated code. https://sites.google.com/site/siddharthamandal1985/research. Accessed 19 Sep 2019.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60.
Huttenhower C. Galaxy /Hutlab—Harvard. http://huttenhower.sph.harvard.edu/galaxy/. Accessed 19 Sep 2019.
beta-rarefaction: Beta diversity rarefaction—QIIME 2 2020.8.0 documentation. https://docs.qiime2.org/2020.8/plugins/available/diversity/beta-rarefaction/?highlight=beta%20rarefaction. Accessed 30 Dec 2021.
glm function—RDocumentation. https://www.rdocumentation.org/packages/stats/versions/3.6.2/topics/glm. Accessed 29 Mar 2021.
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, et al. pROC: display and analyze ROC curves. 2021. https://CRAN.R-project.org/package=pROC. Accessed 29 Mar 2021.
Ramsheh MY, Haldar K, Esteve-Codina A, Purser LF, Richardson M, Müller-Quernheim J, et al. Lung microbiome composition and bronchial epithelial gene expression in patients with COPD versus healthy individuals: a bacterial 16S rRNA gene sequencing and host transcriptomic analysis. Lancet Microbe. 2021;2:e300–10.
Russell AB, Wexler AG, Harding BN, Whitney JC, Bohn AJ, Goo YA, et al. A type VI secretion-related pathway in bacteroidetes mediates interbacterial antagonism. Cell Host Microbe. 2014;16:227–36.
Dickson RP, Martinez FJ, Huffnagle GB. the role of the microbiome in exacerbations of chronic lung diseases. Lancet. 2014;384:691–702.
Bouquet J, Tabor DE, Silver JS, Nair V, Tovchigrechko A, Griffin MP, et al. Microbial burden and viral exacerbations in a longitudinal multicenter COPD cohort. Respir Res. 2020;21:77.
Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359:2355–65.
Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS ONE. 2010;5: e8578.
Larsen JM. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology. 2017;151:363–74.
Segal LN, Clemente JC, Tsay J-CJ, Koralov SB, Keller BC, Wu BG, et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol. 2016;1:16031.
Larsen JM, Steen-Jensen DB, Laursen JM, Søndergaard JN, Musavian HS, Butt TM, et al. Divergent pro-inflammatory profile of human dendritic cells in response to commensal and pathogenic bacteria associated with the airway microbiota. PLoS ONE. 2012;7: e31976.
Haldar K, George L, Wang Z, Mistry V, Ramsheh MY, Free RC, et al. The sputum microbiome is distinct between COPD and health, independent of smoking history. Respir Res. 2020;21:183.
Mosca A, Leclerc M, Hugot JP. Gut microbiota diversity and human diseases: should we reintroduce key predators in our ecosystem? Front Microbiol. 2016. https://doi.org/10.3389/fmicb.2016.00455/abstract.
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–30.
de SteenhuijsenPiters WAA, Sanders EAM, Bogaert D. The role of the local microbial ecosystem in respiratory health and disease. Phil Trans R Soc B. 2015;370:20140294.
Lundmark A, Hu YOO, Huss M, Johannsen G, Andersson AF, Yucel-Lindberg T. Identification of salivary microbiota and its association with host inflammatory mediators in periodontitis. Front Cell Infect Microbiol. 2019;9:216.
Si Y, Fan H, Song Y, Zhou X, Zhang J, Wang Z. Association between periodontitis and chronic obstructive pulmonary disease in a chinese population. J Periodontol. 2012;83:1288–96.
Lin M, Li X, Wang J, Cheng C, Zhang T, Han X, et al. Saliva microbiome changes in patients with periodontitis with and without chronic obstructive pulmonary disease. Front Cell Infect Microbiol. 2020;10:124.
Acknowledgements
We are grateful to all people with COPD and healthy individuals who accepted to participate in this study and to the team of Respiratory Research and Rehabilitation Laboratory, School of Health Sciences (Lab3R-ESSUA) for their support.
Funding
This study is integrated in “GENIAL—Genetic and Clinical markers of COPD trajectory”, “PRIME—Pulmonary Rehabilitation and microbiota in exacerbations of COPD” and "MicroAgeing—The role of microbiota in ageing", funded by Programa Operacional de Competitividade e Internacionalização—COMPETE, through Fundo Europeu de Desenvolvimento Regional—FEDER (POCI-010145-FEDER-028806 and POCI-01-0145-FEDER-007628), Fundação para a Ciência e Tecnologia—FCT (PTDC/DTP-PIC/2284/2014, PTDC/SAU-SER/28806/2017 and PTDC/BIA-EVL/30212/2017) and under the project UIDB/04501/2020. S. Melo-Dias was supported by Grant SFRH/BD/140908/2018 from FCT. A. Sousa was funded from national funds through FCT—Fundação para a Ciência e a Tecnologia, I.P., under the Scientific Employment Stimulus—Institutional Call—reference CEECINST/00026/2018.
Author information
Authors and Affiliations
Contributions
SM-D and AS had full access to all data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. AM was responsible for ethical approval, AM and AS were responsible for obtaining the funding, conception and design of the study. CV and LA provided a substantial contribution for data acquisition. SM-D conducted sample processing and DNA extraction procedures. SMD, and AS conducted Bioinformatics and statistical analysis. SM-D and AS drafted the manuscript. All authors critically revised the manuscript, ensured accuracy and integrity of the work. Al authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
A cross-sectional study was conducted. Ethical approvals were obtained from Administração Regional de Saúde Centro (64/2016) and from Centro Hospitalar do Baixo Vouga (08-03-17). Written informed consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
S. Melo-Dias has nothing to disclose. C. Valente has nothing to disclose. L. Andrade has nothing to disclose. A. Marques has nothing to disclose. A. Sousa has nothing to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of the article was revised: The Supplementary 2 file has been updated.
Supplementary Information
Additional file 1:
Table S1. Sociodemographic, anthropometric and clinical database of study participants.
Additional file 2: Supplementary Methods. Table S2.
Summary of clinical parameters distribution across Clusters I and II. Comparisons between clusters were conducted with Mann-Whitney U-test and chi-square test. Table S3. Summary table of significant logistic regression models established for both GOLD D and hospital admission, adjusted for Pack-years. Coefficients were represented in model equations. Figure S1. Salivary bacteria composition is different between patients with COPD and healthy controls. A) Bar-plot representing the differentially abundant genera between moderate patients with COPD and healthy controls inferred by LEfSe at a significance cut-off of 3. B) Bar-plot representing the differentially abundant genera between moderate patients with COPD and healthy controls inferred by LEfSe at a significance cut-off of 3. C) Bar-plot representing the differentially abundant genera between severe patients with COPD and healthy controls inferred by LEfSe at a significance cut-off of 3. A), B) and C) Differentially abundant OTUs inferred by ANCOM at 0.7 significance cut-off are underlined. Figure S2. Salivary bacteria composition is different between the two clusters. Bar-plot representing the differentially abundant genera between cluster I and cluster II inferred by LEfSe at a significance cut-off of 3. Differentially abundant OTUs inferred by ANCOM at 0.7 significance cut-off are underlined.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Melo-Dias, S., Valente, C., Andrade, L. et al. Saliva as a non-invasive specimen for COPD assessment. Respir Res 23, 16 (2022). https://doi.org/10.1186/s12931-022-01935-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-022-01935-9