
Abstract
Background
Platelet count is a prognostic indicator in the general population and elderly. Thrombocytosis during acute exacerbation of COPD (AECOPD) has been associated with mortality; however, the relationship between platelet count and mortality in stable COPD is unknown.
Methods
We performed post hoc secondary analysis on a subsample of 1797 patients in the Study to Understand Mortality and Morbidity in COPD (SUMMIT) who had blood samples drawn at baseline. Participants were current or former smokers, 40–80 years old with moderate COPD and history or increased risk of cardiovascular (CV) disease. The primary outcome was on and post-treatment all-cause mortality. Secondary outcomes included first-on-treatment moderate/severe AECOPD and on-treatment CV composite event (CV death, myocardial infarction, stroke, unstable angina and transient ischemic attack). Multivariable Cox proportional hazards models were used to investigate study endpoint associations with platelet count quintile grouping, continuous platelet count utilizing two-term fractional polynomials, and categories of low, normal and high platelet count (< 150, ≥150 to < 300, ≥300 × 109/L).
Results
Patients were followed for 2.3 ± 0.9 years for vital status and 1.6 ± 1.1 years for morbidity endpoints during which 105 (5.8%) died, 651 (36.2%) experienced AECOPD (159 with severe AECOPD) and 86 (4.8%) experienced a CV event. A U-shaped association between platelet count and all-cause mortality was observed. Compared to the third quintile group (Q3) of platelet count, risk of death was increased in the lowest quintile group (Q1; hazard ratio [HR]: 1.73; 95% confidence interval [CI]: 0.93–3.23) and highest quintile group (Q5; HR: 1.66; 95%CI: 0.89–3.10), though point estimates were imprecise. Using clinical cutoffs, compared with normal platelet counts (≥150 to < 300 × 109/L), risk of all-cause mortality was nominally increased among patients with thrombocytopenia (HR: 1.46; 95%CI: 0.81–2.64) and high platelet count (HR: 1.66; 95%CI: 0.96–2.86). Compared with Q3, CV events were nominally increased for Q5 (HR: 1.71; 95%CI: 0.83–3.49) and Q1 (HR: 1.41; 95%CI: 0.70, 2.85). There was no association between platelet count and AECOPD.
Conclusions
In stable COPD platelet count demonstrated a U-shaped association with increased risk of 3-year all-cause mortality, though a platelet count level above or below which risk of mortality was increased could not be definitively identified.
Trial registration
ClinicalTrials.gov NCT01313676.
Similar content being viewed by others


Background
Chronic obstructive pulmonary disease (COPD) is projected to become the third leading cause of death by 2030, representing 8.6% of global deaths [1]. Chronic systemic inflammation in COPD has been associated with poor clinical outcomes [2, 3]. Biomarkers have potential value as prognostic indicators in COPD, though their utility has been limited by disease heterogeneity and the influence of comorbidities [4]. Platelet count, a component of a routinely measured clinical assay, remains within a relatively narrow range among healthy individuals but can be significantly altered in the setting of both acute and chronic disease [5]. Notably, platelet count is correlated with levels of fibrinogen [6], an inflammatory biomarker identified as a means of selecting research participants with COPD at high risk for morbidity and mortality [7, 8]. A U-shaped association between platelet count and increased mortality has been recognized in the general population and the elderly [9,10,11,12,13,14], though its role in COPD remains unclear. In addition to the well documented role of platelets in cardiovascular (CV) disease, platelets are now recognized as playing a role in many pathophysiologic processes such as inflammation, host defense, and tumor biology [15].
Overall, platelet count in COPD has been shown to be elevated compared with the general population [16, 17]. Previous studies have demonstrated an increased risk of in-hospital and 1-year mortality associated with thrombocytosis (defined as platelet count > 400 × 109/L) measured at the time of acute exacerbation of COPD (AECOPD) [18]. In univariate analysis, thrombocytopenia measured on admission for AECOPD was also associated with in-hospital mortality, need for mechanical ventilation, intensive care unit admission, and length of hospitalization [19]. Our group has recently demonstrated that elevated platelet count (> 350 × 109/L) measured in stable COPD is associated with higher likelihood of prior exacerbation and worse respiratory symptoms [20]. However, the relationship between platelet count measured in the stable phase of COPD and all-cause mortality has not been previously explored. Furthermore, comorbid COPD and CV disease has been associated with higher rates of morbidity than presence of either disease alone making this a particularly vulnerable population [21]. Using data from the Study to Understand Mortality and Morbidity in COPD (SUMMIT), a prospective randomized controlled trial enriched for presence of CV comorbidity and containing adjudicated events, we investigated post hoc the association of platelet count measured in the stable phase of COPD with all-cause mortality, CV and respiratory outcomes.
Methods
Study population
SUMMIT was a prospective, multi-center, international randomized controlled trial to determine whether treatment with an inhaled long-acting beta-agonist (LABA) in combination with an inhaled corticosteroid (ICS), versus either component, could improve clinical outcomes in patients with moderate COPD and increased CV risk compared with placebo. Details regarding study design have been previously published [22, 23]. In brief, participants were required to be current or former smokers (≥10 pack-years) between the ages of 40 and 80 years, with a history of moderate COPD defined as post-bronchodilator forced expiratory volume in 1 s (FEV1) between 50 and 70% of the predicted value, a ratio of post-bronchodilator FEV1 to forced vital capacity ≤0.70, and a score ≥ 2 on the modified Medical Research Council (mMRC) dyspnea scale. Patients were additionally required to have a history, or be at increased risk, of CV disease. CV disease was defined as coronary artery disease, peripheral arterial disease, prior stroke or myocardial infarction, or diabetes mellitus with target organ disease. Increased CV risk was defined as being ≥60 years and receiving medications for two or more of the following: hypercholesterolemia, hypertension, diabetes mellitus or peripheral vascular disease.
While prior ICS and LABA treatments were discontinued before study entry, other COPD medications were permitted during the trial. Participants were then allocated equally to one of four randomized treatments: placebo, fluticasone furoate (100 μg), vilanterol (25 μg) or their combination (fluticasone furoate/vilanterol, 100/25 μg) inhaled once daily as a dry powder. A total of 16,485 patients were enrolled and included in the final intention-to-treat efficacy population. This secondary analysis included a subsample of 1797 patients who had blood samples drawn, all of whom were recruited in the United States.
Platelet count
Venous blood samples were obtained before randomization and at 3 months. Blood was processed, and plasma stored at − 80 °C until analyzed. Platelet counts were performed by Q2 Solutions (Valencia, CA). Only baseline platelet count was used in this analysis to avoid survivorship bias and ensure that platelet count is not affected by competing outcomes.
Outcomes
The primary outcome of interest is on- and post-treatment all-cause mortality, with secondary outcomes including first-on-treatment AECOPD or on-treatment CV event defined as a composite of CV death, myocardial infarction, stroke, unstable angina and transient ischemic attack [24]. Moderate AECOPD was defined as a symptomatic deterioration requiring treatment with antibiotic drugs or systemic corticosteroids, whereas severe AECOPD was defined as an event leading to hospital admission. Categorization of the cause of each death was adjudicated by a clinical endpoint committee blinded to the treatment allocation who also determined whether any reported CV event met the definition of the composite endpoint [22]. Follow-up visits were not performed after a patient discontinued study treatments therefore the only complete data for those patients was regarding mortality. Data on CV events and COPD exacerbations were no longer collected.
Statistical analysis
Patients were split into quintile groups based on their baseline platelet count and patient characteristics were summarized.
To explore the effect of baseline platelet count quintile on each of the study endpoints (all-cause mortality, CV composite, moderate/severe AECOPD and severe AECOPD), analysis of time-to-first event was performed using Cox proportional hazards regression modelling, using the middle quintile group as the reference. In the mortality analysis participants who did not die were censored at the end of the study, or when they were lost to follow-up if this occurred first. For all other analyses patients were censored if they discontinued use of study treatments or were lost to follow-up. Models were adjusted for age, sex, race, ethnicity, BMI category, smoking status, percent predicted FEV1, previous history of AECOPD, anemia (hematocrit < 36% for women and < 39% for men), SUMMIT CV entry criteria (aged < 60 with CV disease, aged ≥60 with CV disease, aged ≥60 with CV risk), ischemic and vascular disease indicators (e.g., previous treatment of coronary or vascular disease), mMRC score, and study treatment. Sensitivity analysis including use of antiplatelet therapy as a covariate was performed for all outcomes.
Baseline platelet count was also examined as a continuous variable where the best fitting model was selected from a variety of polynomial or logarithmic models using two-term fractional polynomials [25]. As a secondary analysis, patients were regrouped into three categories of low, normal and high platelet count (< 150, ≥150 to < 300, and ≥ 300 × 109/L) based on a standard clinical definition of thrombocytopenia and a platelet count threshold above which previous reports in the general population have identified an increased mortality risk [9, 11,12,13].
Statistical analyses were performed using SAS 9.4 (Carey, NC). All patients provided written informed consent. The study was approved by local ethics committees and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Scientific oversight of the trial was provided by a steering committee composed of academic experts and employees from GlaxoSmithKline plc, who were collectively responsible for the study design and analysis, and for the review and interpretation of the data. This study is registered with ClinicalTrials.gov, number NCT01313676.
Results
Among the subgroup of 1797 participants with platelet count measurement median platelet count was 219 × 109/L (interquartile range [IQR] 181–260 × 109/L). One participant with platelet count 1415 × 109/L was excluded from analysis. Baseline characteristics are presented in Table 1 for the entire cohort and by platelet count quintile group (Q1: < 173 × 109; Q2: ≥173 to < 205 × 109; Q3: ≥205 to < 234 × 109; Q4: ≥234 to < 272 × 109; Q5: ≥272 × 109). The higher platelet count groups included more females of younger average age with lower prevalence of obesity and cardiac comorbidities but higher prevalence of anemia and current smoking.
Patients were on study treatment for an average of 1.6 (± 1.1 SD) years for a total of 3848 person-years, during which time 86 (4.8%) experienced a CV event and 651 (36.2%) experienced an AECOPD, of which 159 (24.4%) subjects experienced a severe AECOPD. Patients were followed-up on and post treatment for an average of 2.3 ± 0.9 years for vital status during which time 105 (5.8%) participants died (Table 2). Four participants were lost to follow-up and did not have vital status recorded at the end of the study.
Participants in the highest quintile group of platelet count had the highest proportion of moderate/severe AECOPD (41%) and severe AECOPD (11%). CV composite events (6%) and deaths (8%) in the highest quintile were equivalent to the lowest quintile of platelet count but higher than the middle quintiles.
Association of platelet count with mortality
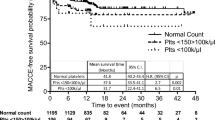
In multivariable analysis a U-shaped association between platelet count and all-cause mortality was observed (Fig. 1). Compared with the third quintile (Q3) of platelet count, patients in the lowest quintile (Q1) had a 73% increased risk of death (95% confidence interval [CI]: − 7 to 223%) while patients in the highest quintile (Q5) had a 66% increased risk of death (95% CI: -11 to 210%), though these estimates were imprecise. Cause-specific mortality by platelet count quintile is presented in Table 2.

Association of platelet count with death from any cause. Open circles and 95% confidence intervals represent hazard ratio in each quintile in reference to middle quintile, shown as a filled circle. Curve and shaded region show hazard ratio and 95% confidence region of platelet count as a continuous variable in reference to the median platelet count
When using clinical ranges, compared with patients who had platelet counts of 150-300 × 109/L, those with thrombocytopenia (< 150 × 109/L) had a 46% increased risk of death (95% CI: -19 to 164%) and those with high platelet count (≥300 × 109/L) had a 66% increased risk (95% CI: -4 to 186%); however, similar to quintile analyses, point estimates were imprecise.
Association of platelet count with CV and respiratory endpoints
Higher platelet counts also were associated with nominally increased hazard of a CV composite event when analyzed as platelet count quintiles (Q5 vs. Q3 hazard ratio [HR]: 1.71; 95% CI: 0.83–2.49). Analyses using clinical ranges showed that, compared with normal platelet count (≥150 to < 300 × 109/L), elevated platelet count (≥300 × 109/L) was associated with 55% increased risk of composite CV outcome (95% CI: -18 to 194%), though this association was also not statistically significant. Compared with Q3, patients in Q1 had nominally increased composite CV events (HR: 1.41; 95% CI: 0.70, 2.85), though this was not consistent with results of secondary analyses (Fig. 2, Table 3). There was no significant association between platelet count and AECOPD, both moderate/severe and severe (Fig. 2, Tables 2 and 3). Sensitivity analyses including use of antiplatelet therapy as an additional covariate produced equivalent results for all outcomes (data not shown).

Association of platelet count with first on-treatment (a) moderate/severe COPD exacerbations, b severe COPD exacerbations, and (c) cardiovascular composite event (cardiovascular death, myocardial infarction, stroke, unstable angina, and transient ischemic attack) Open circles and 95% confidence intervals represent hazard ratio in each quintile in reference to middle quintile, shown as a filled circle. Curve and shaded region show hazard ratio and 95% confidence region of platelet count as a continuous variable in reference to the median platelet count
Discussion
In this prospective study of patients with COPD and prior history or increased risk of CV disease platelet count measured in the stable state demonstrated a U-shaped association with increased risk of 3-year all-cause mortality, though a platelet count level above or below which risk of mortality was increased could not be definitively identified. Higher platelet count also showed trends towards increased risk for a composite outcome of CV morbidity and mortality though these results did not reach statistical significance. Platelet count did not appear to be associated with prospective risk for AECOPD. To our knowledge, this is the first study investigating the association between platelet count measured in the stable state of COPD and all-cause mortality. These findings expand upon prior studies reporting increased all-cause mortality among COPD patients with thrombocytosis and thrombocytopenia measured during an AECOPD [18, 19].
The U-shaped association between platelet count and mortality observed in this sample of stable COPD patients has been previously described in a general adult population [13, 14], postmenopausal women [9], and the elderly [10,11,12]. Increased risk of all-cause mortality was consistently noted in these prior studies for platelet count > 300 × 109/L but not thrombocytopenia (< 150 × 109/L) [11,12,13]. While higher platelet counts appeared to be associated with increased mortality in this study of COPD patients, platelet count ≥300 × 109/L and thrombocytopenia trended toward increased risk of mortality, though did not reach statistical significance. This may be a consequence of a relatively small overall and group specific sample sizes compared with studies of non-COPD cohorts or risk for mortality present only at higher platelet counts, similar to those identified in prior studies of mortality following AECOPD (> 400 × 109/L) and respiratory symptoms (≥350 × 109/L, which we did not have adequate power to evaluate [18, 20].
Elevated platelet count has been associated with CV mortality in healthy middle-aged men but not the elderly [11, 12, 26], and with a composite CV outcome of myocardial infarction, cerebrovascular disease, or peripheral vascular disease in the general population [13]. Though we were not able to evaluate CV mortality independently due to low event rate, higher platelet count, evaluated as a continuous variable, appeared associated with a composite CV outcome in this sample of COPD patients with history or increased risk of CV disease, though higher platelet count categories did not reach statistical significance. Notably, elevated platelet count was not associated with AECOPD in this study which is in contrast to a prior study demonstrating association between platelet count ≥350 × 109/L measured in the stable state and report of at least one AECOPD in the prior year [20]. Thus, it appears that platelet count may not inform risk of future AECOPD beyond prior exacerbation history. Additionally, in this higher risk sample of stable COPD patients with comorbid CV disease, platelet count appears to be a predictor of CV rather than respiratory morbidity.
Potential biological mechanisms for the association of elevated platelet count with all-cause mortality and CV morbidity in COPD include systemic inflammation, atherosclerotic plaque destabilization, and platelet activation. Previous studies investigating the prognostic value of biomarkers among patients with COPD inform the interpretation of the current findings. Fibrinogen, an acute phase protein elevated in systemic inflammation, has demonstrated an association with all-cause mortality in COPD [7]. A strong association between fibrinogen levels and platelet count has been reported [6]. Interleukin-6, a pro-inflammatory cytokine which has also been associated with increased mortality in COPD [27, 28], induces fibrinogen gene expression [29] and increases thrombopoetin production, which increases circulating platelets [30]. Several inflammatory mediators are known to activate platelets, which can then bind to vascular endothelial cells or adherent leukocytes, perpetuating or destabilizing a plaque [31]. The receptor for one inflammatory mediator, platelet activating factor, which perpetuates platelet activation and is associated with CV morbidity, is upregulated within airway epithelial cells of individuals with COPD suggesting a potential role in COPD with comorbid CV disease [32, 33]. Furthermore, in a cohort of individuals with COPD and stable coronary artery disease undergoing percutaneous coronary intervention, dual antiplatelet therapy with Ticagrelor improved markers of endothelial dysfunction and platelet activation suggesting that these mechanisms may be involved in the morbidity of coincident COPD and CV disease that was observed in this study [34].
A high prevalence of cancer related mortality was present in the lowest platelet count quintile, despite exclusion of patients with known malignancy from SUMMIT, which suggests presence of an underlying comorbidity with high cancer risk, subclinical malignancy or bone marrow suppression that may be driving increased mortality risk in this group. An association between thrombocytopenia and increased cancer mortality has been consistently reported in prior studies [9, 11,12,13]. Thrombocytopenia is also associated with increased mortality risk when measured at the time of hospital admission for community acquired pneumonia, independent of infection severity [35]. In this population of COPD patients at high risk for respiratory infections, thrombocytopenia in the stable state likely places them at higher risk of thrombocytopenia and poor outcomes during future acute illness.
There are limitations to this study. Few patients were represented in the lowest and highest platelet count groups, which limited the power to make inferences about these groups. The infrequency of mortality events precluded statistical comparison of case-specific mortality between platelet count quintiles. The study sample included moderate COPD patients with CV disease history or risk factors, which may limit generalizability to all COPD patients. Finally, inflammatory biomarkers were not measured in this study, which precluded investigation of correlations between platelet count and inflammatory biomarkers and limited inferences regarding mechanism.
Conclusions
In conclusion, among patients with moderate COPD and CV disease history or risk factors, there is a U-shaped association between platelet count and all-cause mortality. Higher platelet count was also associated with trends towards increased risk of composite CV disease, although effect estimates for platelet count quintiles and categories were imprecise for all contrasts. Platelet count measured at baseline did not predict future COPD exacerbations.
Abbreviations
- AECOPD:
-
Acute exacerbation of chronic obstructive pulmonary disease
- COPD:
-
Chronic obstructive pulmonary disease
CV
Cardiovascular
- FEV1 :
-
Forced expiratory volume in 1 s
- ICS:
-
Inhaled corticosteroid
- LABA:
-
Long-acting beta-agonist
- mMRC:
-
modified Medical Research Council
- Q1:
-
First/lowest quintile
- Q3:
-
Third/middle quintile
- Q5:
-
Fifth/highest quintile
- SUMMIT:
-
Study to Understand Mortality and Morbidity in COPD
References
WHO. World health statistics 2008. Geneva: World Health Organization; 2008.
Oudijk ED, Lammers JJ, Koenderman L. Systemic inflammation in chronic obstructive pulmonary disease. Eur Respir J. 2003;22(46 suppl):5s–13s.
Agustí A, Edwards LD, Rennard SI, et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One. 2012;7(5):e37483.
Lacoma A, Prat C, Andreo F, Dominguez J. Biomarkers in the management of COPD. Eur Respir Rev. 2009;18(112):96–104.
Duke W. Variation in the platelet count: its cause and clinical significance. J Am Med Assoc. 1915;65(19):1600–7.
Aliberti G, Proietta M, Pulignano I, Del Porto F, Tammeo A, Trappolini M. Association between fibrinogen plasma levels and platelet counts in an outpatient population and in patients with coronary heart disease. Blood Coagul Fibrinolysis. 2010;21(3):216–20.
Mannino DM, Tal-Singer R, Lomas DA, et al. Plasma fibrinogen as a biomarker for mortality and hospitalized exacerbations in people with COPD. Chron Obstruct Pulmon Dis. 2015;2(1):23.
Casaburi R, Celli B, Crapo J, et al. The COPD biomarker qualification consortium (CBQC). COPD: J Chron Obstruct Pulmon Dis. 2013;10(3):367–77.
Kabat GC, Kim MY, Verma AK, et al. Platelet count and total and cause-specific mortality in the Women's Health Initiative. Ann Epidemiol. 2017;27(4):274–80.
Msaouel P, Lam AP, Gundabolu K, et al. Abnormal platelet count is an independent predictor of mortality in the elderly and is influenced by ethnicity. Haematologica. 2014;99(5):930–6.
Tsai M-T, Chen Y-T, Lin C-H, Huang T-P, Tarng D-C. U-shaped mortality curve associated with platelet count among older people: a community-based cohort study. Blood. 2015;126(13):1633–5.
van der Bom JG, Heckbert SR, Lumley T, et al. Platelet count and the risk for thrombosis and death in the elderly. J Thromb Haemost. 2009;7(3):399–405.
Vinholt PJ, Hvas A-M, Frederiksen H, Bathum L, Jørgensen MK, Nybo M. Platelet count is associated with cardiovascular disease, cancer and mortality: a population-based cohort study. Thromb Res. 2016;148:136–42.
Bonaccio M, Di Castelnuovo A, Costanzo S, et al. Age-sex–specific ranges of platelet count and all-cause mortality: prospective findings from the MOLI-SANI study. Blood. 2016;127(12):1614–6.
Ghoshal K, Bhattacharyya M. Overview of platelet physiology: its hemostatic and nonhemostatic role in disease pathogenesis. TheScientificWorldJournal. 2014;2014:781857.
Biljak VR, Pancirov D, Cepelak I, Popovic-Grle S, Stjepanovic G, Grubisic TZ. Platelet count, mean platelet volume and smoking status in stable chronic obstructive pulmonary disease. Platelets. 2011;22(6):466–70.
Wang RT, Li JY, Cao ZG, Li Y. Mean platelet volume is decreased during an acute exacerbation of chronic obstructive pulmonary disease. Respirology. 2013;18(8):1244–8.
Harrison MT, Short P, Williamson PA, Singanayagam A, Chalmers JD, Schembri S. Thrombocytosis is associated with increased short and long term mortality after exacerbation of chronic obstructive pulmonary disease: a role for antiplatelet therapy? Thorax. 2014;69(7):609–15.
Rahimi-Rad MH, Soltani S, Rabieepour M, Rahimirad S. Thrombocytopenia as a marker of outcome in patients with acute exacerbation of chronic obstructive pulmonary disease. Adv Respir Med. 2015;83(5):348–51.
Fawzy A, Putcha N, Paulin LM, et al. Association of thrombocytosis with COPD morbidity: the SPIROMICS and COPDGene cohorts. Respir Res. 2018;19(1):20.
Bafadhel M, Russell RE. Are COPD and cardiovascular disease fundamentally intertwined? Eur respiratory Soc. 2016;47(5):1307–9.
Vestbo J, Anderson J, Brook RD, Calverley PM, Celli BR, Crim C, Haumann B, Martinez FJ, Yates J, Newby DE. The Study to Understand Mortality and Morbidity in COPD (SUMMIT) study protocol. Eur Respir J. 2013;41(5):1017–22.
Vestbo J, Anderson JA, Brook RD, et al. Fluticasone furoate and vilanterol and survival in chronic obstructive pulmonary disease with heightened cardiovascular risk (SUMMIT): a double-blind randomised controlled trial. Lancet. 2016;387(10030):1817–26.
Brook RD, Anderson JA, Calverley PM, et al. Cardiovascular outcomes with an inhaled beta2-agonist/corticosteroid in patients with COPD at high cardiovascular risk. Heart. 2017;103(19):1536–42.
Royston P, Sauerbrei W. A new approach to modelling interactions between treatment and continuous covariates in clinical trials by using fractional polynomials. Stat Med. 2004;23(16):2509–25.
Thaulow E, Erikssen J, Sandvik L, Stormorken H, Cohn PF. Blood platelet count and function are related to total and cardiovascular death in apparently healthy men. Circulation. 1991;84(2):613–7.
Ferrari R, Tanni SE, Caram LM, Corrêa C, Corrêa CR, Godoy I. Three-year follow-up of interleukin 6 and C-reactive protein in chronic obstructive pulmonary disease. Respir Res. 2013;14(1):24.
Celli BR, Locantore N, Yates J, et al. Inflammatory biomarkers improve clinical prediction of mortality in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185(10):1065–72.
Fuller GM, Zhang Z. Transcriptional control mechanism of fibrinogen gene expression. Ann NY Acad Sci. 2001;936(1):469–79.
Schafer AI. Thrombocytosis. N Engl J Med. 2004;350(12):1211–9.
Stokes KY, Granger DN. Platelets: a critical link between inflammation and microvascular dysfunction. J Physiol. 2012;590(5):1023–34.
Shukla SD, Sohal SS, Mahmood MQ, Reid D, Muller HK, Walters EH. Airway epithelial platelet-activating factor receptor expression is markedly upregulated in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2014;9:853.
Palur Ramakrishnan AVK, Varghese TP, Vanapalli S, Nair NK, Mingate MD. Platelet activating factor: a potential biomarker in acute coronary syndrome? Cardiovasc Ther. 2017;35(1):64–70.
Campo G, Dalla Sega FV, Pavasini R, et al. Biological effects of ticagrelor over clopidogrel in patients with stable coronary artery disease and chronic obstructive pulmonary disease. Thromb Haemost. 2017;117(06):1208–16.
Mirsaeidi M, Peyrani P, Aliberti S, et al. Thrombocytopenia and thrombocytosis at time of hospitalization predict mortality in patients with community-acquired pneumonia. Chest. 2010;137(2):416–20.
Acknowledgements
The authors would like to thank the SUMMIT steering committee [22], composed of academic experts and employees from GlaxoSmithKline plc, who were responsible for the scientific oversight, study design and analysis and review and interpretation of the SUMMIT Study.
Funding
NIH NIEHS F32ES028576 (AF); GlaxoSmithKline plc (SUMMIT; HZC113782/NCT01313676).
Availability of data and materials
Information on GlaxoSmithKline plc’s data sharing commitments and requesting access to anonymized individual participant data and associated documents can be found at www.clinicalstudydatarequest.com.
Author information
Authors and Affiliations
Contributions
During the development of the manuscript, AF, NNH and NJC were involved in data analysis/interpretation; CC, JAA, and JCY were involved in the study conception/design, the acquisition of data, and data analysis/interpretation; RW was involved in the study conception/design, and data analysis/interpretation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by local ethics committees and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
Consent for publication
Not applicable.
Competing interests
AF declares no conflict of interest. JAA, CC and JCY are employees of, and own shares in, GlaxoSmithKline plc. NJC is employed by Veramed Ltd., a Contract Research Organisation undertaking contracted statistical analyses of respiratory studies funded by GlaxoSmithKline plc. RW served as the Chair of the Summit Clinical Endpoint Committee and has received grant support and received consulting fees from GlaxoSmithKline plc during the conduct of the study. He has received grants and consulting fees from AstraZeneca and Boehringer Ingelheim. He has received consulting fees from Contrafect, Pulmonx, Roche, Spiration, Sunovion, Merck, Circassia, Verona, Mylan, Theravance, and Propeller Health unrelated to the submitted work. NNH has participated on advisory boards for GlaxoSmithKline plc, Mylan and AstraZeneca, and has received Research Grants from GlaxoSmithKline plc, AstraZeneca and Boehringer Ingelheim.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fawzy, A., Anderson, J.A., Cowans, N.J. et al. Association of platelet count with all-cause mortality and risk of cardiovascular and respiratory morbidity in stable COPD. Respir Res 20, 86 (2019). https://doi.org/10.1186/s12931-019-1059-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-019-1059-1