
Abstract
Background
Asthmatics that are exposed to inhaled pollutants such as cigarette smoke (CS) have increased symptom severity. Approximately 25% of adult asthmatics are thought to be active smokers and many sufferers, especially in the third world, are exposed to high levels of inhaled pollutants. The mechanism by which CS or other airborne pollutants alter the disease phenotype and the effectiveness of treatment in asthma is not known. The aim of this study was to determine the impact of CS exposure on the phenotype and treatment sensitivity of rodent models of allergic asthma.
Methods
Models of allergic asthma were configured that mimicked aspects of the asthma phenotype and the effect of CS exposure investigated. In some experiments, treatment with gold standard asthma therapies was investigated and end-points such as airway cellular burden, late asthmatic response (LAR) and airway hyper-Reactivity (AHR) assessed.
Results
CS co-exposure caused an increase in the LAR but interestingly attenuated the AHR. The effectiveness of LABA, LAMA and glucocorticoid treatment on LAR appeared to be retained in the CS-exposed model system. The eosinophilia or lymphocyte burden was not altered by CS co-exposure, nor did CS appear to alter the effectiveness of glucocorticoid treatment. Steroids, however failed to reduce the neutrophilic inflammation in sensitized mice exposed to CS.
Conclusions
These model data have certain parallels with clinical findings in asthmatics, where CS exposure did not impact the anti-inflammatory efficacy of steroids but attenuated AHR and enhanced symptoms such as the bronchospasm associated with the LAR. These model systems may be utilised to investigate how CS and other airborne pollutants impact the asthma phenotype; providing the opportunity to identify novel targets.
Similar content being viewed by others


Background
Asthma is a respiratory disease that is increasing in prevalence globally. Airborne pollutants such as cigarette smoke (CS, direct and passive) and traffic/industrial pollution are reported to increase asthma susceptibility, cause quality of life issues and enhance symptom severity, frequency of attacks and disease exacerbations [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23]. Smoking and passive smoking has also been shown to adversely impact on the effectiveness of standard treatment such as inhaled corticosteroid (ICS) in asthmatics [24,25,26,27,28] and worsen disease outcome [29]. Despite the fact that asthma is a severe and debilitating illness, a significant proportion of asthma patients smoke or are exposed to passive smoke [30]. As many as half of all adult asthma patients may be active, or previous smokers [13, 14]. Thus with the increase in airborne pollution levels, especially in developing countries, and continued exposure to CS (either directly or passively), it is important to try and understand the mechanism by which these pollutants impact on asthma pathogenesis and whether this contributes to treatment-resistance.
Within allergic asthma, exposure to allergen results in a biphasic bronchoconstrictor response. Immediate bronchoconstriction as a result of exposure is termed the Early Asthmatic Response (EAR) and typically occurs within 1 h of contact with aeroallergen. The Late Asthmatic Response (LAR) refers to a more prolonged bronchoconstriction event taking place approximately 3–8 h following contact with allergen. The LAR is often used within clinical studies exploring new therapeutic options with which to treat asthma and as such is considered to be a clinically relevant endpoint [11, 31].
Airway Hyper-Responsivity (AHR) is a cardinal feature of the asthma phenotype. It is defined as an increased sensitivity to inhaled stimuli resulting in narrowing of the airways, which would not usually occur in healthy individuals. This response manifests as excessive bronchoconstriction and airflow limitation, resulting in shortness of breath and chest tightness. Stimuli of AHR include pollution, allergens, cold air and spasmogens such as Methacholine (MCh). The endpoint of AHR in allergic asthmatics exposed to CS has been investigated but results are sparse and conflicting.
Many features of allergic asthma have been successfully modeled in rats and mice. The Brown Norway rat is considered to be one of the most suitable rat strain for use as an allergic asthma model. This particular strain is a high IgE producer, it produces robust responses to allergens (distinct EAR and LAR) and the infiltration of allergic airway inflammation is considered to be similar to that seen in asthmatic patients [31, 32]. The mouse is also considered to be an advantageous model of allergic asthma due to the possibility of the application of genetically modified (GM) strains and the fact that it comprises a highly characterised immune system.
The aim of this study was to determine the effect of CS co-exposure on the phenotype and treatment sensitivity of rodent models of allergic asthma. In order to investigate this, rodent models of allergic asthma were co-exposed to CS and endpoints of the Late Asthmatic Response (LAR), Airway Hyper-Responsivity (AHR) and airway cellular burden were assessed. The effectiveness of gold standard asthma treatments (i.e. ICS, LABA and LAMA) were also investigated within these models. It was hypothesised that the allergic asthma models exposed to CS would exhibit enhanced LAR and AHR responses and the efficacy of standard asthma treatments would be diminished within these groups.
Methods
Animals
All experimental protocols were approved by a local ethical review process and strictly adhered to the Animals (Scientific Procedures) Act 1986 UK Home Office guidelines and performed according to the ARRIVE guidelines. Male Brown Norway rats (200-250 g) and male C57BL/6 mice were obtained from Harlan, UK. All animals were housed in individually ventilated cages (IVC) and a 12-h light-dark cycle maintained. Prior to and during experimental periods, food and water was supplied ad libitum.
Cigarette smoke exposure system
CS exposure was performed according to methods as previously described by our laboratory [33, 34]. Briefly, filtered research cigarettes (University of Kentucky Research Cigarettes, [Ref: #3R4F]) were stored at 4 °C, and 48 h prior to use they were brought to room temperature and the filters removed. On the day of exposures, the exposure system equipment was set up as previously described [33, 34] and the system flow set to 1.5 L/min. Animals were placed in stainless steel cages (rats: 12 per cage; mice: 40 per cage) before being placed into the system. Cigarettes were administered to the system via the pinch valve. Smoke exposure sessions lasted for 50 mins, defined as the last cigarette being removed from the system 50 mins from the first being lit. The system flow was checked at 15 min intervals, and the wellbeing of the animals checked continually through each session with the use of a torch for visibility. Total Suspended Particulate (TSP) was sampled at 30 min intervals in each exposure session. A filter membrane was weighed prior to being administered to the dry gas meter sampling unit. Each TSP sample period was 1 min. The filter was weighed again at the end of the sampling period. The dry gas meter recordings were noted at the start and end of each exposure period. TSP was calculated as follows:
The TSP values were consistent and as such a consistent CS burden could be confirmed.
Investigating the effect of cigarette smoke exposure in the Brown Norway rat model of the LAR
A rat model of allergic asthma was used as previously described [32]. Briefly, male Brown Norway rats were sensitised on day 0, 14 and 21 with chicken ovalbumin (OVA) (100 μg/rat, i.p., Grade V, Sigma, UK.) administered with Alum (20 mg/rat aluminium hydroxide and 20 mg/rat magnesium hydroxide, i.p., Alum™ Thermo Scientific, UK). Rats were exposed to room air or CS for 1 h, twice a day (4 h apart) on day 21, 22, 23, 24, 25, 26, and 27. On day 28 the rats were exposed to air/smoke in the morning and in the afternoon the rats were challenged with vehicle (saline, aerosolised for 30 min) or OVA (1% w/v, aerosolised for 30 min). The LAR was monitored in conscious BN rats for 1 to 6 h after challenge as previously described [31, 35]. The following day the animals were euthanised with pentobarbitone (200 mg/kg, i.p., Centaur Services, UK). Bronchoalveolar lavage (BAL) was carried out by injecting 3 ml of RPMI culture medium (Invitrogen, UK) via a cannula inserted into the trachea, waiting 30 s and then removing it. This was repeated and the collected BAL fluid (BALF) pooled. Total and type of white cells in the BALF were determined as previously described [32, 35].
Assessing the effectiveness of standard asthma therapies in the CS co-exposed rat model of the LAR
To determine if CS co-exposure alters the effectiveness of current asthma therapies, rats were treated with topical glucocorticoid, budesonide; LABA, Olodaterol, and LAMA, glycopyrrolate. Briefly, under inhaled anaesthetic, rats (n = 8) received vehicle (0.5% ethanol in saline, 1 ml/kg, intratracheal), Olodaterol (1 mg/kg, dose selected from preliminary studies), budesonide (3 mg/kg, dose selected from previous work [35]) or glycopyrrolate (1 mg/kg, dose selected from preliminary studies) one hour before and 30 min after OVA challenge. LAR was measured in all three studies as described in the previous section and airway inflammation was assessed in the study with glucocorticoid intervention.
Investigating the effect of cigarette smoke exposure in a C57BL/6 model of AHR
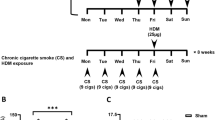
A mouse model that we have previously shown to feature AHR was applied to this body of work [36, 37]. Briefly, male C57bl/6 mice were sensitised on day 0 and 14 with either OVA (10 μg/ mouse, i.p.) with Alum™ (diluted 1:1 with saline, 100 μl i.p.) or HDM (0.5 μg/kg in 100 ul, i.p. from Greer, USA – No Alum™). On days 24, 25, 26 mice were challenged intranasally with vehicle (50 ul saline), OVA (2.5 mg/kg) or HDM (1.25 μg/kg). Purified HDM extract from Dermatophagoides pteronyssinus (Der p; lot number 124632; GREER laboratories, USA) with a known content of Der p1 (12.76 μg/mg dry weight) was used in these experiments. Endotoxin content – 125 EU/vial (121 μg HDM /vial).
Mice were exposed to room air or CS for 1 h, twice a day (4 h apart) on days 21 to 28. This CS exposure protocol was based on previous development work [33] using a system previously described [34]. On day 29 lung function (Penh) was assessed to increasing doses of inhaled spasmogen (aerosolised 5-HT) using a method previously described [36]. After the lung function had returned to pre-spasmogen levels the mice were euthanised with pentobarbitone (200 mg/kg, i.p., Centaur Services, UK). BAL was carried out by injecting 0.3 ml of RPMI culture medium (Invitrogen, UK) via a cannula inserted into the trachea, waiting 30 s and then removing it. This was repeated twice more and the collected BALF pooled. Total and type of white cells in the BALF was determined as previously described [36].
Assessing the effectiveness of steroid asthma therapy in the CS co-exposed mouse model of AHR.
To determine if CS co-exposure alters the effectiveness of glucocorticoids, mice received vehicle (0.5% methylcellulose plus 0.2% tween80 in water, 10 ml/kg, orally) or budesonide (0.3, 1 or 3 mg/kg, orally, doses selected from previous work [36]) twice per day on days 24–28 (1 h prior to the morning CS exposure and 1 h after the afternoon CS exposure). Airway cellular burden was assessed on day 29 as described above. AHR was not assessed because CS co-exposure attenuated the signal.
Data analysis
Data are expressed as mean ± S.E.M. of n observations. Statistical significance was determined using either single or multiple comparisons (specific tests used are described in the Figure legends), using GraphPad Prism 5 software. A P value < 0.05 was taken as significant and all treatments were compared with the appropriate control group.
Results
Effect of cigarette smoke exposure on a rat model of allergic asthma
Antigen challenge led to a marked increase in respiratory distress (increased audible and visual signs) that correlated with a change in Penh levels in sensitised rats, as previously shown and described as a LAR [31, 35] (Fig. 1a). Exposure to CS alone appeared to have no effect compared to the appropriate control group, but when rats were co-exposed with the antigen there was an increase in the magnitude of the LAR (Fig. 1a).

Effect of CS co-exposure on a rat model of allergic asthma. Sensitised male Brown Norway rats were challenged with an aerosol of saline or OVA for 30 min. Rats were co-exposed to room air or CS (twice a day) for eight days. Changes in lung function (Penh) were assessed from 1 h after the end of challenge for 5 h. BALF was collected the following day and the numbers of white cells assessed. Data are represented as mean ± S.E.M. for n = 8 animals in each group. a: LAR, b: neutrophil number, c: Eosinophil number and d: lymphocyte number. The statistical significance of the response to antigen and/or CS was determined using a Mann-Whitney U test and denoted with # (P < 0.05)
The day after antigen challenge, we observed a significant increase in neutrophils, eosinophils and lymphocytes in the BALF (Fig. 1b-d). CS alone caused a small but statistically significant increase in BALF neutrophilia (Fig. 1b), but did not alter the allergic cellular inflammation triggered with antigen challenge (Fig. 1b-d).
To determine if the CS plus antigen challenge phenotype had an altered sensitivity to gold standard asthma treatment we profiled a topical glucocorticoid, LABA and LAMA. Figure 2 shows that antigen challenge increased the BALF levels of eosinophils, neutrophils and lymphocytes. CS alone increased neutrophil number but did not significantly alter the response to antigen (Fig. 2). Topical treatment with the clinically relevant glucocorticoid, budesonide, inhibited the cellular inflammation in both the antigen alone and the antigen plus CS co-exposed animals (Fig. 2). This would suggest that co-exposure with CS did not alter the anti-inflammatory effectiveness of budesonide within this model.

Effect of glucocorticoid treatment on a CS co-exposure rat model of allergic asthma. Sensitised male Brown Norway rats were challenged with an aerosol of saline or OVA for 30 min. Rats were co-exposed to room air or CS for eight days and dosed with vehicle (1 ml/kg, intratracheal) or budesonide (3 mg/kg, i.t) one hour prior to and 30 min after antigen challenge. BALF was collected the following day and the numbers of white cells assessed. Data are represented as mean ± S.E.M. for n = 8 animals in each group. a: Eosinophil number, b: neutrophil number and c: lymphocyte number. The statistical significance of the response to antigen and/or CS was determined using Mann-Whitney and denoted with # (P < 0.05). The significance of the impact of budesonide was determined using Mann-Whitney and denoted with * (P < 0.05)
Treatment with topical glucocorticoid, LABA and LAMA impacted on the LAR observed after antigen challenge (Fig. 3). CS exposure alone appeared to have no direct effect on changes in Penh but co-exposure increased the LAR. The CS co-exposed LAR signal was almost completely blocked by treatment with olodaterol or glucocorticoid and attenuated by glycopyrrolate (Fig. 3). This would indicate that although co-exposure with CS leads to an enhanced LAR in this model, this particular asthma phenotype is still sensitive to topical glucocorticoid and bronchodilator treatment.

Effect of gold standard asthma treatments on a CS co-exposure rat model of allergic asthma. Sensitised male Brown Norway rats were challenged with an aerosol of saline or OVA for 30 min. Rats were co-exposed to room air or CS for eight days and dosed with vehicle (1 ml/kg, intratracheal, i.t.), budesonide (3 mg/kg, i.t), Olodaterol (1 mg/kg, i.t.) or glycopyrrolate (1 mg/kg, i.t.) one hour prior to and 30 min after antigen challenge. Changes in lung function were assessed from 1 h after the end of challenge for 5 h. Data are represented as mean ± S.E.M. for n = 8 animals in each group. a: Glucocortoid, b: LABA and c: LAMA
Effect of cigarette smoke exposure on mouse models of allergic asthma
Exposure to antigen, either OVA or HDM, resulted in AHR to the inhaled spasmogen (5-HT) (Fig. 4a and b). Intriguingly, whilst exposure to CS alone did not appear to alter responses to 5-HT, in both model systems CS co-exposure attenuated the AHR. Antigen challenge caused a significant increase in BAL eosinophils, neutrophils and lymphocytes (Fig. 4c, d and e). CS alone significantly increased neutrophil number in the BAL but did not alter the level of eosinophils and lymphocytes in BAL after HDM challenge. An additive effect was observed for the neutrophilic inflammation after combined HDM and CS challenge (Fig. 4).

Effect of CS co-exposure on mouse models of allergic asthma. Sensitised male mice were challenged with intranasal saline (50 ul) or antigen (OVA or HDM) once a day for 3 days. Mice were co-exposed to room air or CS for eight days. Changes in airway reactivity (AR) to inhaled 5-HT were assessed 3 days after the final antigen challenge. BALF was then collected and the numbers of white cells assessed. Data are represented as mean ± S.E.M. for n = 8 animals in each group. a: AR after OVA challenge, b: AR after HDM c: Eosinophil number, d: Neutrophil number and e: lymphocyte number. The statistical significance of the response to antigen and/or CS was determined using a Mann-Whitney U test and denoted with # (P < 0.05)
As CS co-exposure attenuated AHR in both model systems, we could not determine the impact of standard asthma therapies on this end point. Therefore we profiled the anti-inflammatory effects of a glucocorticoid (budesonide, administered p.o.) on the cellular inflammation only. As can be seen in Fig. 5, antigen challenge increased BAL cellular inflammation, and this signal was inhibited by treatment with budesonide. Co-exposure to CS did not appear to impact on the effectiveness of budesonide treatment on antigen induced increase in eosinophil and lymphocyte numbers (Fig. 5). Neutrophil numbers after CS challenge alone, or in combination with antigen challenge, was not altered by budesonide treatment as previously reported [38].

Effect of glucocorticoid treatment on CS co-exposure mouse models of allergic asthma. Sensitised male mice were challenged with intranasal saline (50 ul) or antigen (OVA or HDM) once a day for 3 days. Mice were co-exposed to room air or CS for eight days. BALF was then collected and the numbers of white cells assessed. Mice were dosed with vehicle (10 ml/kg, orally, p.o.) or budesonide (3 mg/kg, p.o.) one hour prior to the morning CS challenges and one hour after afternoon CS challenges. Data are represented as mean ± S.E.M. for n = 8 animals in each group. a: Eosinophil number, b: Neutrophil number and c: lymphocyte number. The statistical significance of the response to antigen and/or CS was determined using a Mann-Whitney U test and denoted with * (P < 0.05). The significance of the impact of budesonide was determined using one way ANOVA followed by a Bonferoni’s correction post-test # (P < 0.05)
Discussion
Airborne pollutants such as CS (direct and passive) are known to increase asthma symptoms, severity, frequency of attacks and disease exacerbations and to adversely impact the effectiveness of standard treatment such as inhaled corticosteroid (ICS) in asthmatics. Despite this, the levels of smoking in asthmatic patients are still high; with some estimates suggesting that smoking asthmatics in developed countries represent approximately one quarter of all sufferers. Thus it is important to try and understand the mechanism by which pollution impacts on asthma pathogenesis and treatment. To investigate this effect we determined how CS altered the asthma phenotype in rodent models of allergic asthma. Our studies showed that CS co-exposure increased the magnitude of the LAR, but actually inhibited the AHR signal. CS co-exposure did not appear to impact on cellular burden (above and beyond an additive effect) or treatment effectiveness. This is the first pre-clinical study to comprehensively examine the impact of CS co-exposure on the asthmatic phenotype, and the data demonstrates that these models have many parallels with clinical observations suggesting their usefulness for future investigations.
Antigen challenge triggered cellular recruitment in sensitised animals as previously reported [35, 39]. Similarly exposure to CS caused the expected increase in airway neutrophilia [40]. Co-exposure of the allergic asthma models with CS appeared not to alter the cellular profile above and beyond an additive effect (i.e. neutrophil number). Similar increases in neutrophil numbers have been reported in asthmatics that smoke [41, 42] and it is believed that this cell type plays an important role in the pathophysiology of asthma and is linked to the “asthma COPD overlap syndrome”. Furthermore, Meghji et al. have recently shown similar eosinophilia data in human asthmatics demonstrating that smoking status does not alter the levels following antigen challenge [43]. Interestingly there are some reports that eosinophil numbers are reduced in asthmatics that smoke [14, 44]. This observation could depend on a number of factors including the level of smoke exposure/pack years, asthmatic status and time of sampling. The published preclinical data from studies examining the effect of CS co-exposure is varied, with some reporting reductions and others augmentation in cellular inflammation (the main focus is often eosinophil numbers) [45,46,47,48,49,50,51,52,53,54,55,56,57,58,59]. These disparate findings appear to be largely due to variations in CS co-exposure protocols.
Treatment with a clinically relevant corticosteroid, budesonide, inhibited the allergen induced cellular inflammation in the model systems as expected [35, 36], whilst it failed to impact on the CS induced neutrophilia as previously shown [38, 60, 61]. In our studies, co-exposure with CS did not appear to impact the effectiveness of budesonide treatment, a similar result was published by Song et al. [59]. Surprisingly few clinical studies have described the effects of steroid treatment on airway inflammation in smoking asthmatics; the studies tend to report lung function or asthma control as the primary endpoint. In addition, if pulmonary cellular inflammation is described, it is typically only eosinophilia that is reported, therefore there is little direct evidence on the effects of steroids on other inflammatory cells in smoking asthmatics. ICS have been shown to reduce sputum eosinophils in asthmatics, but not in smoking asthmatics in short term and long term studies [62], but others have shown that ICS do improve sputum eosinophils and ECP in smokers and non-smokers alike [29]. Therefore, the effect of smoking on the anti-inflammatory effects of steroids in asthmatics is currently controversial.
A striking observation is the apparent blockade of AHR in the model systems, whether it was driven by an allergic response to OVA or HDM. A similar finding was recently reported in asthmatic smokers that were exposed to a range of antigens and challenged with inhaled MCh [43]. As stated by the authors, it is not clear what the clinical significance is of this observation. One could speculate that as it is well known that smoking does increase clinical symptoms, the measurement of airway reactivity could be clinically irrelevant. Another group has reported that smoke challenge increases AHR in asthmatics but these experiments were performed using a sub-population of asthmatics that have previously reported to be sensitive to CS [22, 23]. Furthermore, the change was observed in only 30% of this sub-population and a similar number were affected in non-asthmatics. Other pre-clinical studies have reported similar findings with CS co-exposure inhibiting the AHR [54, 55]. Currently the mechanism by which CS causes this effect is not known. Melgert et al. (2004) suggested it was through the reduction of cellular inflammation in their model, but this seems unlikely as in our model systems since cellular inflammation was not decreased. There has been some speculation as to whether CS could be directly or indirectly evoking bronchodilation. Indeed CS is known to contain carbon monoxide which has been reported to reduce mouse AHR [63]; furthermore, CS can induce the release of bronchodilation substances such as PGE2 and nitric oxide [64]. In addition, CS contains nicotine, which conceivably could alter AHR. We believe, however, that these mechanisms are unlikely as normal airway reactivity to inhaled spasmogen was not altered by CS exposure, and the model systems presented with a strong LAR signal. Both these end points should be altered if CS was causing bronchodilation. Other possible mechanisms by which CS co-exposure reduces the AHR signal could be through the reduction of the mediators driving the AHR and the many cytokines suggested to be involved such as IL-5, IL-13 and IL-17 [65,66,67,68,69,70,71] or the production of mediators reported to inhibit AHR like TGFb [72, 73]. Indeed it has been reported that CS co-exposure increases levels of TGFb [74]. Unfortunately measurement of these end points is not possible in our studies as they were designed to focus on cellular inflammation and AHR, and not cytokine levels (the optimum time for cytokine measurements is much earlier) [75]. Another possible mechanism by which CS alters AHR could be due to an impact on airway smooth muscle (ASM), either the increased ability to contract [76] or the remodelling changes reported such as increased ASM thickness via antigen induced increase in proliferation/migration associated with the AHR phenotype [77]. Of the published studies, some have suggested CS increases proliferation, some have suggested inhibition and others to modulate the contractile response, thus this mechanism is still a possibility but needs to be further investigated [78,79,80,81,82,83,84,85,86]. Finally, CS could be causing remodelling in the airway which subsequently impacts on AHR. Indeed it has been reported that CS increases airway remodelling in pre-clinical asthma models [48, 52, 87].
Despite the loss of the AHR phenotype in the models following CS exposure, the LAR remains a clear feature; a similar observation was made in smoking human asthmatics [43]. Indeed, our data suggests that CS co-exposure actually enhances this cardinal feature of asthma. It is therefore tempting to speculate that it is this symptom of asthma that is central to the detrimental impact CS has on asthmatics.
As far as we know, we are the first to examine the effect of CS co-exposure on the LAR in a preclinical model. It is currently not clear how CS is exacerbating the LAR signal. One could speculate that as previous data has strongly implicated the TRPA1 - sensory nerve – parasympathetic axis in the LAR [31] that CS is somehow modulating elements of this pathway. Indeed it is well known that CS contains elements like acrolein which can activate TRPA1 [88, 89]. Further, TRPA1 is the molecular target for by-products of oxidative stress including Reactive Oxygen Species (ROS) and other electrophilic compounds, including hypochlorite and hydrogen peroxide which are linked to CS exposure [90,91,92,93,94]. As CS alone did not cause a “LAR” like response, it would seem that CS induced exacerbation of the response is not simply due to an increase of TRPA1 activator(s). One possible reason for the synergy between CS and antigen challenge could be that CS is increasing the sensitivity of airway sensory nerves to TRPA1 activators. Indeed we, and others, have observed that CS exposure can increase sensory nerve responses to TRPV1 ligands [95], furthermore we have unpublished data that suggests that TRPA1 responses are also increased. It is interesting to note that whilst we do not yet know the mechanism by which CS exacerbates LAR, current therapies such as ICS and LABA can combat this symptom of asthma. Furthermore, the inhibition of LAR in this model with glycopyrrolate confirms previous finding using another LAMA, tiotropium [31, 96].
Conclusion
The aim our investigation was to determine the effect of CS co-exposure on the phenotype and treatment sensitivity of rodent models of allergic asthma. In order to investigate this, rodent models of allergic asthma were co-exposed to CS and endpoints of the Late Asthmatic Response (LAR), Airway Hyper-Responsivity (AHR) and airway cellular burden were assessed. The impact of ICS, LAMA and LABA were also observed within these models.
In summary, we found that the magnitude of LAR within the allergen sensitised models increased with co-exposure to CS and is concordant with our initial hypothesis. Divergent with our hypothesis; ICS, LAMA and LABA attenuated the LAR across both CS exposed and non-exposed groups. Interestingly the AHR was attenuated with exposure to CS. This was accompanied by an increase in neutrophilic inflammation, and although ICS was successful in attenuating overall cellular inflammation, the enhanced neutrophil populations observed remained undiminished.
We suggest that the data from these studies have parallels with clinical findings and that these model systems may be useful tools in helping to understand how exposure to airborne pollutants such as CS can alter the asthmatic phenotype. We propose that these model systems will be extremely useful in future research and will provide the opportunity to identify novel targets for asthma.
Abbreviations
- AHR:
-
Airway hyperresponsiveness
- Alum:
-
20 mg/ml aluminium hydroxide and 20 mg/ml magnesium hydroxide
- AUC:
-
Area under the curve
- BALF:
-
Bronchoalveolar lavage fluid
- i.p.:
-
Intraperitoneal
- i.t.:
-
Intratracheal
- IgE:
-
Immunoglobulin E
- LAR:
-
Late asthmatic response
- OVA:
-
Ovalbumin
- PBS:
-
Phosphate buffered saline
- Penh:
-
Enhanced pause
- S.E.M.:
-
Standard error of the mean
- WBP:
-
Whole body plethysmograph
References
Zmirou D, Gauvin S, Pin I, et al. Traffic related air pollution and incidence of childhood asthma: results of the Vesta case-control study. J Epidemiol Community Health. 2004;58:18–23.
McConnell R, Berhane K, Yao L, et al. Traffic, susceptibility, and childhood asthma. Environ Health Perspect. 2006;114:766–72.
Salam MT, Islam T, Gilliland FD. Recent evidence for adverse effects of residential proximity to traffic sources on asthma. Curr Opin Pulm Med. 2008;14:3–8. https://doi.org/10.1097/MCP.0b013e3282f1987a.
Patel MM, Quinn JW, Jung KH, et al. Traffic density and stationary sources of air pollution associated with wheeze, asthma, and immunoglobulin E from birth to age 5 years among new York City children. Environ Res. 2011;111:1222–9. https://doi.org/10.1016/j.envres.2011.08.004.
Clark NA, Demers PA, Karr CJ, et al. Effect of early life exposure to air pollution on development of childhood asthma. Environ Health Perspect. 2010;118:284–90. https://doi.org/10.1289/ehp.0900916.
Gowers AM, Cullinan P, Ayres JG, et al. Does outdoor air pollution induce new cases of asthma? Biological plausibility and evidence; a review. Respirology. 2012;17:887–98. https://doi.org/10.1111/j.1440-1843.2012.02195.x.
Chung KF, Zhang J, Zhong N. Outdoor air pollution and respiratory health in Asia. Respirology. 2011;16:1023–6. https://doi.org/10.1111/j.1440-1843.2011.02034.x.
Apostol GG, Jacobs DRJ, Tsai AW, et al. Early life factors contribute to the decrease in lung function between ages 18 and 40: the coronary artery risk development in young adults study. Am J Respir Crit Care Med. 2002;166:166–72.
Eisner MD, Iribarren C. The influence of cigarette smoking on adult asthma outcomes. Nicotine Tob Res. 2007;9:53–6. https://doi.org/10.1080/14622200601078293.
Jang A-S, Park J-S, Lee J-H, et al. The impact of smoking on clinical and therapeutic effects in asthmatics. J Korean Med Sci. 2009;24:209–14. https://doi.org/10.3346/jkms.2009.24.2.209.
O’Byrne PM, Lamm CJ, Busse WW, et al. The effects of inhaled budesonide on lung function in smokers and nonsmokers with mild persistent asthma. Chest. 2009;136:1514–20. https://doi.org/10.1378/chest.09-1049.
Siroux V, Pin I, Oryszczyn M, et al. Relationships of active smoking to asthma and asthma severity in the EGEA study. Epidemiological study on the genetics and environment of asthma. Eur Respir J. 2000;15:470–7.
Thomson NC, Chaudhuri R, Livingston E. Asthma and cigarette smoking. Eur Respir J. 2004;24:822–33. https://doi.org/10.1183/09031936.04.00039004.
Thomson NC, Chaudhuri R, Heaney LG, et al. Clinical outcomes and inflammatory biomarkers in current smokers and exsmokers with severe asthma. J Allergy Clin Immunol Published Online First: February 2013. https://doi.org/10.1016/j.jaci.2012.12.1574.
Polosa R, Russo C, Caponnetto P, et al. Greater severity of new onset asthma in allergic subjects who smoke: a 10-year longitudinal study. Respir Res. 2011;12:16. https://doi.org/10.1186/1465-9921-12-16.
Eisner MD, Klein J, Hammond SK, et al. Directly measured second hand smoke exposure and asthma health outcomes. Thorax. 2005;60:814–21. https://doi.org/10.1136/thx.2004.037283.
Eisner MD, Yelin EH, Henke J, et al. Environmental tobacco smoke and adult asthma. The impact of changing exposure status on health outcomes. Am J Respir Crit Care Med. 1998;158:170–5. https://doi.org/10.1164/ajrccm.158.1.9801028.
Stankus RP, Menon PK, Rando RJ, et al. Cigarette smoke-sensitive asthma: challenge studies. J Allergy Clin Immunol. 1988;82:331–8.
Althuis MD, Sexton M, Prybylski D. Cigarette smoking and asthma symptom severity among adult asthmatics. J Asthma. 1999;36:257–64.
Gallefoss F, Bakke PS. Does smoking affect the outcome of patient education and self-management in asthmatics? Patient Educ Couns. 2003;49:91–7.
Dahms TE, Bolin JF, Slavin RG. Passive smoking. Effects on bronchial asthma. Chest. 1981;80:530–4.
Menon PK, Stankus RP, Rando RJ, et al. Asthmatic responses to passive cigarette smoke: persistence of reactivity and effect of medications. J Allergy Clin Immunol. 1991;88:861–9.
Menon P, Rando RJ, Stankus RP, et al. Passive cigarette smoke-challenge studies: increase in bronchial hyperreactivity. J Allergy Clin Immunol. 1992;89:560–6.
Chalmers GW, Macleod KJ, Little SA, et al. Influence of cigarette smoking on inhaled corticosteroid treatment in mild asthma. Thorax. 2002;57:226–30.
Chaudhuri R, Livingston E, McMahon AD, et al. Cigarette smoking impairs the therapeutic response to oral corticosteroids in chronic asthma. Am J Respir Crit Care Med. 2003;168:1308–11. https://doi.org/10.1164/rccm.200304-503OC.
Laforest L, Van Ganse E, Devouassoux G, et al. Influence of patients’ characteristics and disease management on asthma control. J Allergy Clin Immunol. 2006;117:1404–10. https://doi.org/10.1016/j.jaci.2006.03.007.
Chaudhuri R, Livingston E, McMahon AD, et al. Effects of smoking cessation on lung function and airway inflammation in smokers with asthma. Am J Respir Crit Care Med. 2006;174:127–33. https://doi.org/10.1164/rccm.200510-1589OC.
Clatworthy J, Price D, Ryan D, et al. The value of self-report assessment of adherence, rhinitis and smoking in relation to asthma control. Prim Care Respir J. 2009;18:300–5. https://doi.org/10.4104/pcrj.2009.00037.
Lazarus SC, Chinchilli VM, Rollings NJ, et al. Smoking affects response to inhaled corticosteroids or leukotriene receptor antagonists in asthma. Am J Respir Crit Care Med. 2007;175:783–90. https://doi.org/10.1164/rccm.200511-1746OC.
Rabe KF, Adachi M, Lai CKW, et al. Worldwide severity and control of asthma in children and adults: the global asthma insights and reality surveys. J Allergy Clin Immunol. 2004;114:40–7. https://doi.org/10.1016/j.jaci.2004.04.042.
Raemdonck K, de Alba J, Birrell M a, et al. A role for sensory nerves in the late asthmatic response. Thorax. 2012;67:19–25. https://doi.org/10.1136/thoraxjnl-2011-200365.
Underwood S, Foster M, Raeburn D, et al. Time-course of antigen-induced airway inflammation in the Guinea-pig and its relationship to airway hyperresponsiveness. Eur Respir J. 1995;8:2104–13.
Belvisi MG, Birrell MA, Khalid S, Wortley MA, Dockry R, Coote J, Holt K, Dubuis E, Kelsall A, Maher SA, Bonvini S, Woodcock A, Smith JA. Neurophenotypes in Airway Diseases. Insights from Translational Cough Studies. Am J Respir Crit Care Med. 2016;193(12):1364–72. https://doi.org/10.1164/rccm.201508-1602OC
Eltom S, Stevenson C, Birrell MA. Cigarette smoke exposure as a model of inflammation associated with COPD. Curr Protoc Pharmacol. 2013;5:5.64.
Birrell MA, Hardaker E, Wong S, et al. Ikappa-B kinase-2 inhibitor blocks inflammation in human airway smooth muscle and a rat model of asthma. Am J Respir Crit Care Med. 2005;172:962–71. https://doi.org/10.1164/rccm.200412-1647OC.
Birrell M a, Battram CH, Woodman P, et al. Dissociation by steroids of eosinophilic inflammation from airway hyperresponsiveness in murine airways. Respir Res. 2003;4:3.
Raemdonck K, Baker K, Dale N, Dubuis E, Shala F, Belvisi MG, Birrell MA. CD4+ and CD8+ T cells play a central role in a HDM driven model of allergic asthma. Respir Res. 2016;17(1):45. https://doi.org/10.1186/s12931-016-0359-y
Rastrick JMD, Stevenson CS, Eltom S, et al. Correction: cigarette smoke induced airway inflammation is independent of NF-κB Signalling. PLoS One. 2013;8 https://doi.org/10.1371/annotation/754d7b19-2dac-479b-a23c-db9fed0431be.
Birrell MA, De Alba J, Catley MC, et al. Liver X receptor agonists increase airway reactivity in a model of asthma via increasing airway smooth muscle growth. J Immunol. 2008;181:4265–71.
Eltom S, Stevenson CS, Rastrick J, et al. P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLoS One. 2011;6:–e24097. https://doi.org/10.1371/journal.pone.0024097.
Boulet L-P, Lemière C, Archambault F, et al. Smoking and asthma: clinical and radiologic features, lung function, and airway inflammation. Chest. 2006;129:661–8. https://doi.org/10.1378/chest.129.3.661.
St-Laurent J, Bergeron C, Pagé N, et al. Influence of smoking on airway inflammation and remodelling in asthma. Clin Exp Allergy. 2008;38:1582–9. https://doi.org/10.1111/j.1365-2222.2008.03032.x.
Meghji Z, Dua B, Watson RM, et al. Allergen inhalation challenge in smoking compared with non-smoking asthmatic subjects. Clin Exp Allergy. 2011;41:1084–90. https://doi.org/10.1111/j.1365-2222.2011.03782.x.
Broekema M, ten Hacken NHT, Volbeda F, et al. Airway epithelial changes in smokers but not in ex-smokers with asthma. Am J Respir Crit Care Med. 2009;180:1170–8. https://doi.org/10.1164/rccm.200906-0828OC.
Moerloose KB, Pauwels RA, Joos GF. Short-term cigarette smoke exposure enhances allergic airway inflammation in mice. Am J Respir Crit Care Med. 2005;172:168–72. https://doi.org/10.1164/rccm.200409-1174OC.
Moerloose KB, Robays LJ, Maes T, et al. Cigarette smoke exposure facilitates allergic sensitization in mice. Respir Res. 2006;7:49. https://doi.org/10.1186/1465-9921-7-49.
Van Hove CL, Moerloose K, Maes T, et al. Cigarette smoke enhances Th-2 driven airway inflammation and delays inhalational tolerance. Respir Res. 2008;9:42. https://doi.org/10.1186/1465-9921-9-42.
Min MG, Song DJ, Miller M, et al. Coexposure to environmental tobacco smoke increases levels of allergen-induced airway remodeling in mice. J Immunol. 2007;178:5321–8.
Seymour BW, Pinkerton KE, Friebertshauser KE, et al. Second-hand smoke is an adjuvant for T helper-2 responses in a murine model of allergy. J Immunol (Baltimore, Md 1950). 1997;159:6169–75.
Rumold R, Jyrala M, Diaz-Sanchez D. Secondhand smoke induces allergic sensitization in mice. J Immunol (Baltimore, Md 1950). 2001;167:4765–70.
Trimble NJ, Botelho FM, Bauer CMT, et al. Adjuvant and anti-inflammatory properties of cigarette smoke in murine allergic airway inflammation. Am J Respir Cell Mol Biol. 2009;40:38–46. https://doi.org/10.1165/rcmb.2008-0107OC.
Botelho FM, Llop-Guevara A, Trimble NJ, et al. Cigarette smoke differentially affects eosinophilia and remodeling in a model of house dust mite asthma. Am J Respir Cell Mol Biol. 2011;45:753–60. https://doi.org/10.1165/rcmb.2010-0404OC.
Melgert BN, Timens W, Kerstjens HA, et al. Effects of 4 months of smoking in mice with ovalbumin-induced airway inflammation. Clin Exp Allergy. 2007;37:1798–808. https://doi.org/10.1111/j.1365-2222.2007.02843.x.
Melgert BN, Postma DS, Geerlings M, et al. Short-term smoke exposure attenuates ovalbumin-induced airway inflammation in allergic mice. Am J Respir Cell Mol Biol. 2004;30:880–5. https://doi.org/10.1165/rcmb.2003-0178OC.
Robbins CS, Pouladi MA, Fattouh R, et al. Mainstream cigarette smoke exposure attenuates airway immune inflammatory responses to surrogate and common environmental allergens in mice, despite evidence of increased systemic sensitization. J Immunol. 2005;175:2834–42.
Thatcher TH, Benson RP, Phipps RP, et al. High-dose but not low-dose mainstream cigarette smoke suppresses allergic airway inflammation by inhibiting T cell function. Am J Physiol Lung Cell Mol Physiol. 2008;295:L412–21. https://doi.org/10.1152/ajplung.00392.2007.
Lanckacker EA, Tournoy KG, Hammad H, et al. Short cigarette smoke exposure facilitates sensitization and asthma development in mice. Eur Respir J Published Online First: August 2012. https://doi.org/10.1183/09031936.00096612.
Maes T, Provoost S, Lanckacker EA, et al. Mouse models to unravel the role of inhaled pollutants on allergic sensitization and airway inflammation. Respir Res. 2010;11:7. https://doi.org/10.1186/1465-9921-11-7.
Song DJ, Min MG, Miller M, et al. Environmental tobacco smoke exposure does not prevent corticosteroids reducing inflammation, remodeling, and airway hyperreactivity in mice exposed to allergen. Am J Physiol Lung Cell Mol Physiol. 2009;297:L380–7. https://doi.org/10.1152/ajplung.90588.2008.
Marwick JA, Kirkham PA, Stevenson CS, et al. Cigarette smoke alters chromatin remodeling and induces Proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol. 2004;31:633–42. https://doi.org/10.1165/rcmb.2004-0006OC.
Marwick JA, Caramori G, Stevenson CS, et al. Inhibition of PI3Kδ restores glucocorticoid function in smoking-induced airway inflammation in mice. Am J Respir Crit Care Med. 2009;179:542–8. https://doi.org/10.1164/rccm.200810-1570OC.
Pedersen B, Dahl R, Karlström R, et al. Eosinophil and neutrophil activity in asthma in a one-year trial with inhaled budesonide. The impact of smoking. Am J Respir Crit Care Med. 1996;153:1519–29.
Ameredes BT, Otterbein LE, Kohut LK, et al. Low-dose carbon monoxide reduces airway hyperresponsiveness in mice. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1270–6. https://doi.org/10.1152/ajplung.00145.2003.
Chang WC, Lee YC, Liu CL, et al. Increased expression of iNOS and c-fos via regulation of protein tyrosine phosphorylation and MEK1/ERK2 proteins in terminal bronchiole lesions in the lungs of rats exposed to cigarette smoke. Arch Toxicol. 2001;75:28–35.
Foster PS, Hogan SP, Ramsay AJ, et al. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201.
Hamelmann E, Oshiba A, Loader J, et al. Antiinterleukin-5 antibody prevents airway hyperresponsiveness in a murine model of airway sensitization. Am J Respir Crit Care Med. 1997;155:819–25.
Hamelmann E, Takeda K, Schwarze J, et al. Development of eosinophilic airway inflammation and airway hyperresponsiveness requires interleukin-5 but not immunoglobulin E or B lymphocytes. Am J Respir Cell Mol Biol. 1999;21:480–9.
Grünig G, Warnock M, Wakil AE, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–3.
Mattes J, Yang M, Siqueira A, et al. IL-13 induces airways hyperreactivity independently of the IL-4R alpha chain in the allergic lung. J Immunol (Baltimore, Md 1950). 2001;167:1683–92.
Barczyk A, Pierzchala W, Sozañska E. Interleukin-17 in sputum correlates with airway hyperresponsiveness to methacholine. Respir Med. 2003;97:726–33.
Barlow JL, Bellosi A, Hardman CS, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. 2012;129:191–8.4. https://doi.org/10.1016/j.jaci.2011.09.041.
Hansen G, McIntire JJ, Yeung VP, et al. CD4(+) T helper cells engineered to produce latent TGF-beta1 reverse allergen-induced airway hyperreactivity and inflammation. J Clin Invest. 2000;105:61–70. https://doi.org/10.1172/JCI7589.
Schramm C, Herz U, Podlech J, et al. TGF-beta regulates airway responses via T cells. J Immunol (Baltimore, Md 1950). 2003;170:1313–9.
Kim DY, Kwon EY, Hong GU, et al. Cigarette smoke exacerbates mouse allergic asthma through Smad proteins expressed in mast cells. Respir Res. 2011;12:49. https://doi.org/10.1186/1465-9921-12-49.
Birrell MA, Wong S, Catley MC, et al. Impact of tobacco-smoke on key signaling pathways in the innate immune response in lung macrophages. J Cell Physiol. 2008;214:27–37. https://doi.org/10.1002/jcp.21158.
Ma X, Cheng Z, Kong H, et al. Changes in biophysical and biochemical properties of single bronchial smooth muscle cells from asthmatic subjects. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1181–9. https://doi.org/10.1152/ajplung.00389.2001.
Ebina M, Takahashi T, Chiba T, et al. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma. A 3-D morphometric study. Am Rev Respir Dis. 1993;148:720–6.
Fang Q, Zhao M, Ren G. Effects of cigarette smoke extract on proliferation and ET-1 release of airway smooth muscle cells. Zhonghua Yi Xue Za Zhi. 1997;77:201–4.
Lin J, Xu Y, Zhang Z, et al. Effect of cigarette smoke extract on the role of protein kinase C in the proliferation of passively sensitized human airway smooth muscle cells. J Huazhong Univ Sci Technolog Med Sci. 2005;25:269–73.
Pera T, Gosens R, Lesterhuis AH, et al. Cigarette smoke and lipopolysaccharide induce a proliferative airway smooth muscle phenotype. Respir Res. 2010;11:48. https://doi.org/10.1186/1465-9921-11-48.
Zhang X-Y, Xu Y-J, Liu X-S, et al. Cigarette smoke extract promotes proliferation of airway smooth muscle cells in asthmatic rats via regulating cyclin D1 expression. Chin Med J. 2010;123:1709–14.
Xu G-N, Yang K, Xu Z-P, et al. Protective effects of anisodamine on cigarette smoke extract-induced airway smooth muscle cell proliferation and tracheal contractility. Toxicol Appl Pharmacol. 2012;262:70–9. https://doi.org/10.1016/j.taap.2012.04.020.
Stavenow L, Falke P, Berglund A. Effects of different injurious stimuli on cell death, proliferation, and collagen secretion by rabbit aortic smooth muscle cells and human umbilical vein endothelial cells in culture. Med Biol. 1983;61:214–8.
Yoon CH, Park H-J, Cho Y-W, et al. Cigarette smoke extract-induced reduction in migration and contraction in normal human bronchial smooth muscle cells. Korean J Physiol Pharmacol. 2011;15:397–403. https://doi.org/10.4196/kjpp.2011.15.6.397.
Zhang W, Case S, Bowler RP, et al. Cigarette smoke modulates PGE2 and host defence against Moraxella catarrhalis infection in human airway epithelial cells. Respirology. 2011;16:508–16.
Sathish V, Vanoosten SK, Miller BS, et al. Brain-derived neurotrophic factor in cigarette smoke-induced airway hyperreactivity. Am J Respir Cell Mol Biol. 2013;48:431–8. https://doi.org/10.1165/rcmb.2012-0129OC.
Cisneros-Lira J, Gaxiola M, Ramos C, et al. Cigarette smoke exposure potentiates bleomycin-induced lung fibrosis in Guinea pigs. Am J Physiol Lung Cell Mol Physiol. 2003;285:L949–56. https://doi.org/10.1152/ajplung.00074.2003.
Andrè E, Campi B. Cigarette smoke–induced neurogenic inflammation is mediated by α, β-unsaturated aldehydes and the TRPA1 receptor in rodents. J Clin Invest. 2008;118:2574–82. https://doi.org/10.1172/JCI34886.2574.
Simon SA, Liedtke W. How irritating: the role of TRPA1 in sensing cigarette smoke and aerogenic oxidants in the airways. J Clin Invest. 2008;118:2383–6.
Bautista DM. Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci. 2005;102:12248–52.
Bessac BF, Sivula M, von Hehn CA, et al. TRPA1 is a major oxidant sensor in murine airway sensory neurons. J Clin Invest. 2008;118:1899–910.
Takahashi N, Kuwaki T, Kiyonaka S, et al. TRPA1 underlies a sensing mechanism for O2. Nat Chem Biol. 2011;7:701–11.
Sawada S, Suehisa H, Yamashita M. Inhalation of corticosteroid and β-agonist for persistent cough following pulmonary resection. Gen Thorac Cardiovasc Surg. 2012;60:285–8.
Andersson DA, Gentry C, Moss S, et al. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci. 2008;28:2485–94. https://doi.org/10.1523/JNEUROSCI.5369-07.2008.
Dubuis E, Wortley MA, Grace MS, et al. Theophylline inhibits the cough reflex through a novel mechanism of action⋆. J Allergy Clin Immunol. 2014:1–11. https://doi.org/10.1016/j.jaci.2013.11.017.
Smit M, Zuidhof AB, Bos SIT, et al. Bronchoprotection by olodaterol is synergistically enhanced by tiotropium in a Guinea pig model of allergic asthma. J Pharmacol Exp Ther. 2014;348:303–10. https://doi.org/10.1124/jpet.113.208439.
Funding
Studies were supported by the Medical Research Council; (MRC, UK) (MR/K020293/1). KB was supported by an MRC studentship.
Availability of data and materials
Please contact author for data requests.
Author information
Authors and Affiliations
Contributions
KB, NM, KR, BD and MAB carried out the studies and participated in the sample analysis. MGB, ATN and MAB conceived the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Belvisi, M.G., Baker, K., Malloy, N. et al. Modelling the asthma phenotype: impact of cigarette smoke exposure. Respir Res 19, 89 (2018). https://doi.org/10.1186/s12931-018-0799-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-018-0799-7