
Abstract
Heme oxygenase-1 (HO-1) is a rate-limiting enzyme catalyzing oxidative degradation of cellular heme to liberate free iron, carbon monoxide (CO) and biliverdin in mammalian cells. In addition to its primary role in heme catabolism, HO-1 exhibits anti-oxidative and anti-inflammatory functions via the actions of biliverdin and CO, respectively. HO-1 is highly induced in various disease states, including cancer. Several lines of evidence have supported the implication of HO-1 in carcinogenesis and tumor progression. HO-1 deficiency in normal cells enhances DNA damage and carcinogenesis. Nevertheless, HO-1 overexpression in cancer cells promotes proliferation and survival. Moreover, HO-1 induces angiogenesis through modulating expression of angiogenic factors. Although HO-1 is an endoplasmic reticulum resident protein, HO-1 nuclear localization is evident in tumor cells of cancer tissues. It has been shown that HO-1 is susceptible to proteolytic cleavage and translocates to nucleus to facilitate tumor growth and invasion independent of its enzymatic activity. HO-1 also impacts cancer progression through modulating tumor microenvironment. This review summarizes the current understanding of the protumorigenic role of HO-1 and its potential as a molecular target for cancer therapy.
Similar content being viewed by others


Review
Heme oxygenase-1 (HO-1) was initially identified as a liver microsomal protein with activity to degrade heme to bilirubin four decades ago [1]. The first HO-1 cDNA was cloned from rat spleen in 1985 [2], and the amino acid sequence revealed a hydrophobic segment located at the carboxyl terminus, which is required for its anchoring to endoplasmic reticulum (ER) with type II transmembrane topology. HO-1 is highly expressed in the organs responsible for degrading senescent red blood cells, including spleen, reticuloendothelial cells of the liver and bone marrow, and HO-1 in macrophages is involved in the recycling of hemoglobin-heme [1]. Although the basal HO-1 expression level in tissues not directly responsible for hemoglobin metabolism is low, HO-1 is highly induced in cells upon exposure to many agents promoting cellular stresses, including heavy metals, endotoxin, cytokines, heme, hypoxia, nitric oxide, and UV irradiation [2-6]. These findings indicate that HO-1 is not only involved in normal physiology but also has a role in pathophysiological states.
The research on HO-1 was rapidly extended in late 1990’s. It has been shown that HO-1 knockout mice develop anemia associated with hepatic and renal iron overload, which leads to oxidative tissue injury and chronic inflammation [7]. Moreover, these mice are more susceptible to ischemic and reperfusion injury [8,9]. A human case of HO-1 deficiency was also reported [10]. This patient suffered from growth retardation with persistent hemolytic anemia and abnormal coagulation/fibrinolysis system associated with persistent vascular injury. These observations provide strong evidence to support the implication of HO-1 in systemic iron homeostasis and stress response. Along with the genetic studies, increasing evidence has accumulated to show that CO and biliverdin, the byproducts derived from heme degradation by HO-1, possess potent anti-inflammatory and antioxidant activities, respectively [11,12]. These findings provoke substantial interest in the cytoprotective functions of HO-1 and its therapeutic potential in treating various disease states associated with inflammation and oxidative stress, such as cardiovascular and pulmonary diseases [13]. Considering that both oxidative stress and inflammation are implicated in tumorigenesis, the role of HO-1 in cancer has also received considerable attention in recent years.
Association between HO-1 gene polymorphism and cancer
Genetic variation is one of the important factors contributing to cancer susceptibility. A (GT)n repeat polymorphism present in the proximal promoter of human HO-1 gene has been shown to influence the transcriptional activation of HO-1 gene [14]. The shorter (GT) repeats is associated with higher transcriptional activity of HO-1 gene in human cells. Several genetic studies have been carried out to assess the association between HO-1 gene polymorphism and the risk of cancer in humans [15-23]. It has been shown that subjects carrying the shorter (GT) repeats have lower risk in oral squamous cell carcinoma [15], lung adenocarcinoma [16], gastric adenocarcinoma [18], breast cancer [19], esophageal squamous cell carcinoma [21], and malignant mesothelioma [23]. However, some reported the higher risk for melanoma [17], gastric cancer [20], and pancreatic cancer [22] in subjects carrying the shorter repeats. The inconsistency is likely caused in part by the differences in the subject ethnicity, sample sizes, disease stages, and cancer types for studies [24]. It is apparent that further studies with large homogeneous patient populations will be needed to validate the association between HO-1 gene polymorphism and human cancer.
Association between HO-1 expression and cancer progression
HO-1 overexpression is commonly seen in human cancers, including prostate [25], renal [26], gastric [27], colon [28], lung [29], thyroid [30], bladder [31], breast [32], oral [33], and glioma [34] cancers. Moreover, the expression level is positively correlated with the disease stage and poor prognosis in patients. Notably, there were studies showing that HO-1 was detected not only in cytoplasm but also in nucleus of tumor cells in prostate [35,36], lung [29,37], and oral [38] cancer tissues. The extent of HO-1 nuclear localization was associated with disease progression and poor prognosis in patients with prostate cancer [36] and oral carcinoma [38]. In addition to the expression in tumor cells, the positive HO-1 immunoreactivity was also detected in stromal compartment, particularly the tumor-associated macrophages [39-41] of cancer tissues, suggesting that HO-1 may impact cancer progression through modulating tumor microenvironment.
Regulation of HO-1 expression in cancer cells
Cancer cells exhibit elevated oxidative stress due to their high metabolic rate. Moreover, they are surrounded by a complex microenvironment and significantly influenced by their interplays with the stromal components, especially the infiltrating inflammatory cells [42]. It is envisioned that the oxidative stress and stimulations by various growth factors and cytokines released from stromal cells are capable of inducing HO-1 gene transcription in tumor cells through activation of various signaling pathways and transcriptional factors, including Nrf2, NF-κB, AP2 and others [43]. Hypoxia has also been shown to induce HO-1 expression [6].
Furthermore, HO-1 gene expression is upregulated by oncogenes, such as Kaposi sarcoma-herpes virus [44] and BCR/ABL kinase [45]. In addition to the regulation at transcriptional level, HO-1 expression is subjected to posttranscriptional regulation. It has been shown that regulation of HO-1 by mir378 is implicated in lung carcinoma growth and metastasis [46]. Downregulation of HO-1 by mir200c enhances the sensitivity of renal carcinoma cells to chemotoxic agents [47]. Moreover, HO-protein is turnovered by ubiquitin-proteasome system [48]. Our group recently demonstrated that HO-1 is a physiological substrate of TRC8, which is an ER-resident E3 ligase associated with hereditary renal cell carcinoma and thyroid cancer [49]. It was shown that the tumor suppressive effect of TRC8 is mediated at least in part via targeting HO-1 for ubiquitination and degradation in cancer cells.
Paradoxical roles of HO-1 in tumorigenesis
Tumorigenesis is a multistep process in which the accumulation of several genomic mutations is required to initiate the transformation of normal cells to become cancer cells. DNA damage caused by the reactive oxygen species (ROS) is a major source of mutation. HO-1 downregulation leads to the increase of ROS and DNA damage in cells [49]. Furthermore, CO improves cell survival post irradiation or genotoxin treatment by inducing DNA repair [50]. Therefore, increase in HO-1 expression prevents DNA damage and the initiation of carcinogenesis in normal cells. However, at late phase of tumorigenesis, HO-1 overexpression promotes cancer cell proliferation and invasiveness [45,49,51-53]. HO-1 protects cancer cells from apoptosis induced by chemotoxic agents or irradiation, suggesting its involvement in therapeutic resistance [54-61]. A recent study showed that CO contributes to the resistance of cancer cells to oxidative stress and chemotoxic agents by inhibiting the heme-containing cystathionine β-synthase, which causes reduced PFKFB3 methylation and shift of glucose metabolism to pentose phosphate pathway, resulting in subsequent increase of NADPH to replenish reduced glutathione [62].
Paradoxically, another study in prostate cancer demonstrated that CO inhibits tumor growth and increases sensitivity to chemotherapy by enhancing metabolic exhaustion [36]. The cause behind the opposite effects of CO observed in these studies is not yet clear. Nevertheless, HO-1 augments angiogenesis in tumor by inducing the expression of angiogenic factors, such as vascular endothelial growth factor (VEGF) [63-65]. Recently, increasing evidence has demonstrated the involvement of Nrf2-mediated transcriptional activation of antioxidant genes in promoting cell transformation and tumorigenesis [66,67]. Mutations in Nrf2 and its inhibitor, KEAP1, have recently been identified in human cancers [68]. As HO-1 is one of the target genes regulated by Nrf2, it is apparent that HO-1-mediated antioxidant response contributes at least in part to the tumorigenic process promoted by Nrf2 activation. Targeting HO-1 has been shown to be an effective approach for hormone-refractory prostate cancer [69] and overcome imatinib resistance in chronic myeloid leukemia [70]. It also increases the sensitivity of hepatoma, urothelial and pancreatic cancers to chemotherapy [55,60,63,71]. Furthermore, HO-1 inhibition is synthetic lethal in fumarate-hydrotase deficient cells associated with hereditary leiomyomatosis and renal-cell cancer [72].
Protumorigenic function of nuclear HO-1
Although HO-1 is an ER-anchored protein, there were reports showing HO-1 localization in other subcellular compartments [73-75]. HO-1 nuclear localization has been seen in fetal lung cells exposed to hyperoxia [76], and in brown adipocytes and astroglial cells during differentiation [77,78]. HO-1 nuclear localization was also evident in the cancer cells of prostate, lung, and oral cancer tissues, and associated with tumor progression [29,35,36,38]. However, the pathophysiological significance of HO-1 nuclear localization and the mechanism involved are not yet fully explored. Early studies have shown that HO-1 is sensitive to proteolytic cleavage [79]. A recent study demonstrated that HO-1 undergoes proteolytic cleavage, which results in the release of a soluble HO-1 with truncation of its C-terminal transmembrane segment from ER membrane and subsequent translocation to nucleus under some stress conditions in vitro [80]. More recently, studies from our group and others have shown that HO-1 is susceptible to intramembrane cleavage mediated by the ER-associated signal peptide peptidase (SPP) [37,81]. We further demonstrated that SPP is highly expressed in lung cancer cells, and correlates with HO-1 nuclear localization in the same lung cancer tissues [37]. Interestingly, nuclear HO-1 promotes tumor growth and invasion independent of its enzymatic activity [37]. The nuclear HO-1 translocation has also been shown to be implicated in imatinib resistance in chronic myeloid leukemia cells [82]. These findings add a new dimension to HO-1-mediated protumorigenic effects. It has been shown that HO-1 nuclear translocation confers protection against oxidative stress in yeast and mammalian cells through activating oxidant responsive transcriptional factors and upregulation of antioxidant genes. [80,83] Since HO-1 does not contain DNA binding domain, whether it can impact the transcription of genes related to cancer progression through interaction with transcriptional factors or other nuclear proteins deserves further investigation.
Impacts of hematopoietic HO-1 on cancer
The immune/inflammatory cells recruited to tumor microenviroment have profound effects on cancer progression by modulating inflammatory response and anti-tumor immunity [42]. HO-1 has been shown to modulate the immune regulatory functions of myeloid cells by suppressing the expression of proinflammatory cytokines, such as tumor necrosis factor-α, but promoting the expression of immunosuppressivecytokine, interleukin-10 (IL-10) [12]. HO-1 promotes inflammation-associated angiogenesis through up-regulating VEGF expression in macrophages [84]. Furthermore, HO-1 expression in myeloid-derived suppressor cells participates in the suppression of alloreactive T cells [85]. Although HO-1 expression in the stromal macrophages has been seen in the cancer tissues [39-41], the impact of HO-1 expression in myeloid cells on cancer progression is less explored. A recent study by Arnold et al. demonstrated that HO-1 expression mediates the immune suppressive function of a stromal macrophage subpopulation expressing fibroblast activation protein-α [86].
By performing the syngeneic tumor graft experiments with wild type and HO-1 +/− mice, we recently demonstrated that the host HO-1 expression did not affect the growth of primary tumor, but significantly enhanced lung metastasis [87]. The involvement of hematopoietic HO-1 in this process was further demonstrated by the bone marrow transplantation experiment. Mechanistically, we found that HO-1 enhances the chmoattractant-induced migration response of myeloid cells, and therefore facilitates the recruitment of myeloid cells to the pulmonary premetastatic niche and the metastatic loci. Moreover, myeloid HO-1-induced expressions of VEGF and IL-10 promoted tumor cell extravasation and STAT3 activation, which are crucial for the survival and successful colonization of tumor cells in metastatic sites.
In addition to the role in innate immunity, HO-1 also participates in the adaptive immune response in tumor microenvironment. There was a study showing that HO-1-specific CD8+ regulatory T cells with immunosuppressive activity is present in the peripheral blood and tumor tissues of patients [88]. Collectively, these findings support that HO-1 can impact cancer progression through modulating tumor microenvironment.
Conclusion
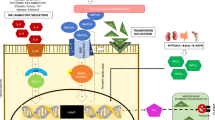
As illustrated in Figure 1, HO-1 can influence cancer development through multiple pathways. HO-1 confers protection in early carcinogenesis, but it promotes cancer cell survival, growth and metastasis in the later process. The paradoxical roles of HO-1 in different phases of tumorigenesis may provide partial explanation for the discrepant findings in the genetic association studies. Beyond its effect on tumor cells, HO-1 can impact cancer progression through modulating tumor microenvironment. Myeloid HO-1 expression promotes the recruitment of immune/inflammatory cells and their immunosuppressive and proangiogenic capacities facilitate cancer cell growth and metastasis. Although considerable protumorigenic effects of HO-1 are mediated by its reaction byproducts, the discovery that HO-1 translocates to nuclear to enhance tumor cell proliferation and invasion via a mechanism independent of its enzymatic activity increases the complexity of HO-1-targeted therapy. Nevertheless, accumulative evidence has demonstrated that HO-1 inhibition using specific gene knockdown approach or metalloprotoporphyrin competitive inhibitor, such as zinc protoporphyrin IX, to block heme binding significantly enhances the sensitivity of cancer cells to chemotherapy or irradiation and suppresses cancer metastasis in experimental animals [55,60,61,63,70.71]. Recently, several imidazole-based non-porphyrin HO-1 inhibitors were developed [89]. These compounds exhibit higher selectivity toward HO-1 without affecting other heme-containing proteins. Moreover, their potent anti-tumor activities have been shown in vitro and in vivo, supporting their therapeutic applications [69,89,90]. Collectively, these findings support the possibility of targeting HO-1 to improve cancer immunotherapy and prevent metastasis, which is the major cause of cancer-associated death.

Multifaceted roles of HO-1 in cancer. HO-1 induction under various cellular stresses impacts tumorigenesis through multiple pathways. CO and biliverdin, the byproducts derived from heme degradation by HO-1 reaction, protect normal cells from transformation in the early phase of tumorigenesis, whereas promote the growth and survival of tumor cells in the late phase of cancer development. Nuclear translocation of HO-1 from ER can affect cancer progression independent of its enzymatic activity. Moreover, HO-1 expression in stromal compartments influences the establishment of cancer permissive microenvironment.
Abbreviations
- CO:
-
Carbon monoxide
- ER:
-
Endoplasmic reticulum
- HO-1:
-
Heme oxygenase-1
- IL-10:
-
Interleukin-10
- ROS:
-
Reactive oxygen species
- VEGF:
-
Vascular endothelial growth factor
References
Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–68.
Alam J, Shibahara S, Smith A. Transcriptional activation of the heme oxygenase gene by heme and cadmium in mouse hepatoma cells. J Biol Chem. 1989;264:6371–5.
Keyse SM, Applegate LA, Tromvoukis Y, Tyrrell RM. Oxidant stress leads to transcriptional activation of the human heme oxygenase gene in cultured skin fibroblasts. Mol Cell Biol. 1990;10:4967–9.
Rizzardini M, Terao M, Falciani F, Cantoni L. Cytokine induction of haem oxygenase mRNA in mouse liver. Interleukin 1 transcriptionally activates the haem oxygenase gene. Biochem J. 1993;290(Pt 2):343–7.
Cantoni L, Rossi C, Rizzardini M, Gadina M, Ghezzi P, Cantoni L, et al. Interleukin-1 and tumour necrosis factor induce hepatic haem oxygenase. Feedback regulation by glucocorticoids. Biochem J. 1991;279(Pt 3):891–4.
Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem. 1997;272:5375–81.
Poss KD, Tonegawa S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci U S A. 1997;94:10925–30.
Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, et al. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest. 1999;103:R23–9.
Liu X, Wei J, Peng DH, Layne MD, Yet SF. Absence of heme oxygenase-1 exacerbates myocardial ischemia/reperfusion injury in diabetic mice. Diabetes. 2005;54:778–84.
Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, et al. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest. 1999;103:129–35.
Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–6.
Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–8.
Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127.
Chen YH, Lin SJ, Lin MW, Tsai HL, Kuo SS, Chen JW, et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Hum Genet. 2002;111:1–8.
Chang KW, Lee TC, Yeh WI, Chung MY, Liu CJ, Chi LY, et al. Polymorphism in heme oxygenase-1 (HO-1) promoter is related to the risk of oral squamous cell carcinoma occurring on male areca chewers. Br J Cancer. 2004;91:1551–5.
Kikuchi A, Yamaya M, Suzuki S, Yasuda H, Kubo H, Nakayama K, et al. Association of susceptibility to the development of lung adenocarcinoma with the heme oxygenase-1 gene promoter polymorphism. Hum Genet. 2005;116:354–60.
Okamoto I, Krogler J, Endler G, Kaufmann S, Mustafa S, Exner M, et al. A microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with risk for melanoma. Int J Cancer. 2006;119:1312–5.
Lo SS, Lin SC, Wu CW, Chen JH, Yeh WI, Chung MY, et al. Heme oxygenase-1 gene promoter polymorphism is associated with risk of gastric adenocarcinoma and lymphovascular tumor invasion. Ann Surg Oncol. 2007;14:2250–6.
Hong CC, Ambrosone CB, Ahn J, Choi JY, McCullough ML, Stevens VL, et al. Genetic variability in iron-related oxidative stress pathways (Nrf2, NQ01, NOS3, and HO-1), iron intake, and risk of postmenopausal breast cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:1784–94.
Sawa T, Mounawar M, Tatemichi M, Gilibert I, Katoh T, Ohshima H. Increased risk of gastric cancer in Japanese subjects is associated with microsatellite polymorphisms in the heme oxygenase-1 and the inducible nitric oxide synthase gene promoters. Cancer Lett. 2008;269:78–84.
Hu JL, Li ZY, Liu W, Zhang RG, Li GL, Wang T, et al. Polymorphism in heme oxygenase-1 (HO-1) promoter and alcohol are related to the risk of esophageal squamous cell carcinoma on Chinese males. Neoplasma. 2010;57:86–92.
Vashist YK, Uzungolu G, Kutup A, Gebauer F, Koenig A, Deutsch L, et al. Heme oxygenase-1 germ line GTn promoter polymorphism is an independent prognosticator of tumor recurrence and survival in pancreatic cancer. J Surg Oncol. 2011;104:305–11.
Murakami A, Fujimori Y, Yoshikawa Y, Yamada S, Tamura K, Hirayama N, et al. Heme oxygenase-1 promoter polymorphism is associated with risk of malignant mesothelioma. Lung. 2012;190:333–7.
Zhang L, Song FF, Huang YB, Zheng H, Song FJ, Chen KX. Association between the (GT)n polymorphism of the HO-1 gene promoter region and cancer risk: a meta-analysis. Asian Pac J Cancer Prev. 2014;15:4617–22.
Maines MD, Abrahamsson PA. Expression of heme oxygenase-1 (HSP32) in human prostate: normal, hyperplastic, and tumor tissue distribution. Urology. 1996;47:727–33.
Goodman AI, Choudhury M, da Silva JL, Schwartzman ML, Abraham NG. Overexpression of the heme oxygenase gene in renal cell carcinoma. Proc Soc Exp Biol Med. 1997;214:54–61.
Yin Y, Liu Q, Wang B, Chen G, Xu L, Zhou H. Expression and function of heme oxygenase-1 in human gastric cancer. Exp Biol Med (Maywood). 2012;237:362–71.
Yin H, Fang J, Liao L, Maeda H, Su Q. Upregulation of heme oxygenase-1 in colorectal cancer patients with increased circulation carbon monoxide levels, potentially affects chemotherapeutic sensitivity. BMC Cancer. 2014;14:436.
Degese MS, Mendizabal JE, Gandini NA, Gutkind JS, Molinolo A, Hewitt SM, et al. Expression of heme oxygenase-1 in non-small cell lung cancer (NSCLC) and its correlation with clinical data. Lung Cancer. 2012;77:168–75.
Wang TY, Liu CL, Chen MJ, Lee JJ, Pun PC, Cheng SP. Expression of heme oxygenase-1 correlates with tumor aggressiveness and BRAF expression in thyroid cancer. Histopathology 2014, Sep 29. doi:10.1111/his.12562.
Miyata Y, Kanda S, Mitsunari K, Asai A, Sakai H. Heme oxygenase-1 expression is associated with tumor aggressiveness and outcomes in patients with bladder cancer: a correlation with smoking intensity. Transl Res. 2014;164:468–76.
Noh SJ, Bae JS, Jamiyandorj U, Park HS, Kwon KS, Jung SH, et al. Expression of nerve growth factor and heme oxygenase-1 predict poor survival of breast carcinoma patients. BMC Cancer. 2013;13:516.
Tsuji MH, Yanagawa T, Iwasa S, Tabuchi K, Onizawa K, Bannai S, et al. Heme oxygenase-1 expression in oral squamous cell carcinoma as involved in lymph node metastasis. Cancer Lett. 1999;138:53–9.
Gandini NA, Fermento ME, Salomon DG, Obiol DJ, Andres NC, Zenklusen JC, et al. Heme oxygenase-1 expression in human gliomas and its correlation with poor prognosis in patients with astrocytoma. Tumour Biol. 2014;35:2803–15.
Sacca P, Meiss R, Casas G, Mazza O, Calvo JC, Navone N, et al. Nuclear translocation of haeme oxygenase-1 is associated to prostate cancer. Br J Cancer. 2007;97:1683–9.
Wegiel B, Gallo D, Csizmadia E, Harris C, Belcher J, Vercellotti GM, et al. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. 2013;73:7009–21.
Hsu FF, Yeh CT, Sun YJ, Chiang MT, Lan WM, Li FA et al. Signal peptide peptidase-mediated nuclear localization of heme oxygenase-1 promotes cancer cell proliferation and invasion independent of its enzymatic activity. Oncogene 2014. doi: 10.1038/onc.2014.166.
Gandini NA, Fermento ME, Salomon DG, Blasco J, Patel V, Gutkind JS, et al. Nuclear localization of heme oxygenase-1 is associated with tumor progression of head and neck squamous cell carcinomas. Exp Mol Pathol. 2012;93:237–45.
Nishie A, Ono M, Shono T, Fukushi J, Otsubo M, Onoue H, et al. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin Cancer Res. 1999;5:1107–13.
Torisu-Itakura H, Furue M, Kuwano M, Ono M. Co-expression of thymidine phosphorylase and heme oxygenase-1 in macrophages in human malignant vertical growth melanomas. Jpn J Cancer Res. 2000;91:906–10.
Boschetto P, Zeni E, Mazzetti L, Miotto D, Lo Cascio N, Maestrelli P, et al. Decreased heme-oxygenase (HO)-1 in the macrophages of non-small cell lung cancer. Lung Cancer. 2008;59:192–7.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37.
Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci U S A. 1994;91:5987–91.
McAllister SC, Hansen SG, Ruhl RA, Raggo CM, DeFilippis VR, Greenspan D, et al. Kaposi sarcoma-associated herpesvirus (KSHV) induces heme oxygenase-1 expression and activity in KSHV-infected endothelial cells. Blood. 2004;103:3465–73.
Mayerhofer M, Florian S, Krauth MT, Aichberger KJ, Bilban M, Marculescu R, et al. Identification of heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in chronic myeloid leukemia. Cancer Res. 2004;64:3148–54.
Skrzypek K, Tertil M, Golda S, Ciesla M, Weglarczyk K, Collet G, et al. Interplay between heme oxygenase-1 and miR-378 affects non-small cell lung carcinoma growth, vascularization, and metastasis. Antioxid Redox Signal. 2013;19:644–60.
Gao C, Peng FH, Peng LK. MiR-200c sensitizes clear-cell renal cell carcinoma cells to sorafenib and imatinib by targeting heme oxygenase-1. Neoplasma. 2014;61:680–9.
Lin PH, Chiang MT, Chau LY. Ubiquitin-proteasome system mediates heme oxygenase-1 degradation through endoplasmic reticulum-associated degradation pathway. Biochim Biophys Acta. 2008;1783:1826–34.
Lin PH, Lan WM, Chau LY. TRC8 suppresses tumorigenesis through targeting heme oxygenase-1 for ubiquitination and degradation. Oncogene. 2013;32:2325–34.
Otterbein LE, Hedblom A, Harris C, Csizmadia E, Gallo D, Wegiel B. Heme oxygenase-1 and carbon monoxide modulate DNA repair through ataxia-telangiectasia mutated (ATM) protein. Proc Natl Acad Sci U S A. 2011;108:14491–6.
Sunamura M, Duda DG, Ghattas MH, Lozonschi L, Motoi F, Yamauchi J, et al. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis. 2003;6:15–24.
Chen GG, Liu ZM, Vlantis AC, Tse GM, Leung BC, van Hasselt CA. Heme oxygenase-1 protects against apoptosis induced by tumor necrosis factor-alpha and cycloheximide in papillary thyroid carcinoma cells. J Cell Biochem. 2004;92:1246–56.
Was H, Cichon T, Smolarczyk R, Rudnicka D, Stopa M, Chevalier C, et al. Overexpression of heme oxygenase-1 in murine melanoma: increased proliferation and viability of tumor cells, decreased survival of mice. Am J Pathol. 2006;169:2181–98.
Liu ZM, Chen GG, Ng EK, Leung WK, Sung JJ, Chung SC. Upregulation of heme oxygenase-1 and p21 confers resistance to apoptosis in human gastric cancer cells. Oncogene. 2004;23:503–13.
Berberat PO, Dambrauskas Z, Gulbinas A, Giese T, Giese N, Kunzli B, et al. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin Cancer Res. 2005;11:3790–8.
Kweon MH, Adhami VM, Lee JS, Mukhtar H. Constitutive overexpression of Nrf2-dependent heme oxygenase-1 in A549 cells contributes to resistance to apoptosis induced by epigallocatechin 3-gallate. J Biol Chem. 2006;281:33761–72.
Kocanova S, Buytaert E, Matroule JY, Piette J, Golab J, de Witte P, et al. Induction of heme-oxygenase 1 requires the p38MAPK and PI3K pathways and suppresses apoptotic cell death following hypericin-mediated photodynamic therapy. Apoptosis. 2007;12:731–41.
Nuhn P, Kunzli BM, Hennig R, Mitkus T, Ramanauskas T, Nobiling R, et al. Heme oxygenase-1 and its metabolites affect pancreatic tumor growth in vivo. Mol Cancer. 2009;8:37.
Furfaro AL, Piras S, Passalacqua M, Domenicotti C, Parodi A, Fenoglio D, et al. HO-1 up-regulation: a key point in high-risk neuroblastoma resistance to bortezomib. Biochim Biophys Acta. 1842;2014:613–22.
Liu YS, Li HS, Qi DF, Zhang J, Jiang XC, Shi K, et al. Zinc protoporphyrin IX enhances chemotherapeutic response of hepatoma cells to cisplatin. World J Gastroenterol. 2014;20:8572–82.
Kongpetch S, Kukongviriyapan V, Prawan A, Senggunprai L, Kukongviriyapan U, Buranrat B. Crucial role of heme oxygenase-1 on the sensitivity of cholangiocarcinoma cells to chemotherapeutic agents. PLoS One. 2012;7:e34994.
Yamamoto T, Takano N, Ishiwata K, Ohmura M, Nagahata Y, Matsuura T, et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat Commun. 2014;5:3480.
Sass G, Leukel P, Schmitz V, Raskopf E, Ocker M, Neureiter D, et al. Inhibition of heme oxygenase 1 expression by small interfering RNA decreases orthotopic tumor growth in livers of mice. Int J Cancer. 2008;123:1269–77.
Miyake M, Fujimoto K, Anai S, Ohnishi S, Kuwada M, Nakai Y, et al. Heme oxygenase-1 promotes angiogenesis in urothelial carcinoma of the urinary bladder. Oncol Rep. 2011;25:653–60.
Birrane G, Li H, Yang S, Tachado SD, Seng S. Cigarette smoke induces nuclear translocation of heme oxygenase 1 (HO-1) in prostate cancer cells: nuclear HO-1 promotes vascular endothelial growth factor secretion. Int J Oncol. 2013;42:1919–28.
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9.
Geismann C, Arlt A, Sebens S, Schafer H. Cytoprotection “gone astray”: Nrf2 and its role in cancer. Onco Targets Ther. 2014;7:1497–518.
Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564–71.
Alaoui-Jamali MA, Bismar TA, Gupta A, Szarek WA, Su J, Song W, et al. A novel experimental heme oxygenase-1-targeted therapy for hormone-refractory prostate cancer. Cancer Res. 2009;69:8017–24.
Mayerhofer M, Gleixner KV, Mayerhofer J, Hoermann G, Jaeger E, Aichberger KJ, et al. Targeting of heat shock protein 32 (Hsp32)/heme oxygenase-1 (HO-1) in leukemic cells in chronic myeloid leukemia: a novel approach to overcome resistance against imatinib. Blood. 2008;111:2200–10.
Miyake M, Fujimoto K, Anai S, Ohnishi S, Nakai Y, Inoue T, et al. Inhibition of heme oxygenase-1 enhances the cytotoxic effect of gemcitabine in urothelial cancer cells. Anticancer Res. 2010;30:2145–52.
Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477:225–8.
Wang XM, Kim HP, Nakahira K, Ryter SW, Choi AM. The heme oxygenase-1/carbon monoxide pathway suppresses TLR4 signaling by regulating the interaction of TLR4 with caveolin-1. J Immunol. 2009;182:3809–18.
Converso DP, Taille C, Carreras MC, Jaitovich A, Poderoso JJ, Boczkowski J. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J. 2006;20:1236–8.
Bindu S, Pal C, Dey S, Goyal M, Alam A, Iqbal MS, et al. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J Biol Chem. 2011;286:39387–402.
Suttner DM, Sridhar K, Lee CS, Tomura T, Hansen TN, Dennery PA. Protective effects of transient HO-1 overexpression on susceptibility to oxygen toxicity in lung cells. Am J Physiol. 1999;276:L443–51.
Giordano A, Nisoli E, Tonello C, Cancello R, Carruba MO, Cinti S. Expression and distribution of heme oxygenase-1 and −2 in rat brown adipose tissue: the modulatory role of the noradrenergic system. FEBS Lett. 2000;487:171–5.
Li Volti G, Ientile R, Abraham NG, Vanella A, Cannavo G, Mazza F, et al. Immunocytochemical localization and expression of heme oxygenase-1 in primary astroglial cell cultures during differentiation: effect of glutamate. Biochem Biophys Res Commun. 2004;315:517–24.
Yoshida T, Ishikawa K, Sato M. Degradation of heme by a soluble peptide of heme oxygenase obtained from rat liver microsomes by mild trypsinization. Eur J Biochem. 1991;199:729–33.
Lin Q, Weis S, Yang G, Weng YH, Helston R, Rish K, et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem. 2007;282:20621–33.
Boname JM, Bloor S, Wandel MP, Nathan JA, Antrobus R, Dingwell KS, et al. Cleavage by signal peptide peptidase is required for the degradation of selected tail-anchored proteins. J Cell Biol. 2014;205:847–62.
Tibullo D, Barbagallo I, Giallongo C, La Cava P, Parrinello N, Vanella L, et al. Nuclear translocation of heme oxygenase-1 confers resistance to imatinib in chronic myeloid leukemia cells. Curr Pharm Des. 2013;19:2765–70.
Collinson EJ, Wimmer-Kleikamp S, Gerega SK, Yang YH, Parish CR, Dawes IW, et al. The yeast homolog of heme oxygenase-1 affords cellular antioxidant protection via the transcriptional regulation of known antioxidant genes. J Biol Chem. 2011;286:2205–14.
Bussolati B, Ahmed A, Pemberton H, Landis RC, Di Carlo F, Haskard DO, et al. Bifunctional role for VEGF-induced heme oxygenase-1 in vivo: induction of angiogenesis and inhibition of leukocytic infiltration. Blood. 2004;103:761–6.
De Wilde V, Van Rompaey N, Hill M, Lebrun JF, Lemaitre P, Lhomme F, et al. Endotoxin-induced myeloid-derived suppressor cells inhibit alloimmune responses via heme oxygenase-1. Am J Transplant. 2009;9:2034–47.
Arnold JN, Magiera L, Kraman M, Fearon DT. Tumoral immune suppression by macrophages expressing fibroblast activation protein-alpha and heme oxygenase-1. Cancer Immunol Res. 2014;2:121–6.
Lin HH, Chiang MT, Chang PC, Chau LY. Myeloid heme oxygenase-1 promotes metastatic tumor colonization in mice. Cancer Sci, In press.
Andersen MH, Sorensen RB, Brimnes MK, Svane IM, Becker JC, Thor Straten P. Identification of heme oxygenase-1-specific regulatory CD8+ T cells in cancer patients. J Clin Invest. 2009;119:2245–56.
Pittala V, Salerno L, Romeo G, Modica MN, Siracusa MA. A focus on heme oxygenase-1 (HO-1) inhibitors. Curr Med Chem. 2013;20:3711–32.
Salerno L, Pittala V, Romeo G, Modica MN, Siracusa MA, Di Giacomo C, et al. Evaluation of novel aryloxyalkyl derivatives of imidazole and 1,2,4-triazole as heme oxygenase-1 (HO-1) inhibitors and their antitumor properties. Bioorg Med Chem. 2013;21:5145–53.
Acknowledgements
This work was partially supported by fundings from Ministry of Science and Technology and Institute of Biomedical Sciences, Academia Sinica, Taiwan ROC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that she has no competing interests.
Authors’ contribution
LYC wrote and approved the manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Chau, LY. Heme oxygenase-1: emerging target of cancer therapy. J Biomed Sci 22, 22 (2015). https://doi.org/10.1186/s12929-015-0128-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-015-0128-0