
Abstract
Background
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematological malignancy. Aberrant expressed genes contribute to the development and progression of T-ALL. However, the regulation underlying their aberrant expression remains elusive. Dysregulated expression of transcription factors and miRNAs played important regulatory roles in the pathogenesis of T-ALL.
Methods
In this study, we analyzed the alteration of transcriptome profiling and regulatory networks between T-ALL sample and normal T cell samples at transcriptional and post-transcriptional levels.
Results
Our results demonstrated that genes related to cell cycle and cell proliferation processes were significantly upregulated in T-ALL comparing to normal samples. Meanwhile, regulatory network analyses revealed that FOXM1, MYB, SOX4 and miR-21/19b as core regulators played vital roles in the development of T-ALL. FOXM1-miR-21-5p-CDC25A and MYB/SOX4-miR-19b-3p-RBBP8 were identified as important feed-forward loops involved in the oncogenesis of T-ALL. Drug-specific analyses showed that GSK-J4 may be an effective drug, and CDC25A/CAPN2/MCM2 could serve as potential therapeutic targets for T-ALL.
Conclusions
This study may provide novel insights for the regulatory mechanisms underlying the development of T-ALL and potential therapeutic targets.
Similar content being viewed by others


Background
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematological malignancy [1], which accounts for 10–15% of pediatric and 25% of adult ALL cases [2]. The anomalous genetic and epigenetic reprogram could arise abnormal proliferation of T lymphoid blasts and transform T-cell precursors into malignant T-ALL lymphoblasts [3]. However, the detail regulatory mechanisms underlying the transformation procedure are still unknown. Thus, a better understanding of the biological regulation involved in the T-ALL pathogenesis is necessary for identifying strategies of prevention and therapy.
Previous studies reported that dysregulation of transcription factors (TFs) to target genes could block myeloid and hematopoietic stem cells differentiation, eventually causing acute leukemia. HOXA9 as an important TF plays vital roles in hematopoietic stem cell expansion and is commonly dysregulated in human acute leukemia [4]. Besides, HOXA9 could co-regulate with CEBPA to suppress the expression of CDKN2A and CDKN2B to increase cell proliferation [5]. MYC is a critical oncogenic TF in T-ALL, the overexpression of MYC and CDKs maintain the leukemic growth by promoting cell proliferation and initiating DNA replication [6]. CHK1 promotes proliferation and viability of T-ALL by down-modulating replication stress and preventing ATM/caspase-3-dependent cell death [7]. Additionally, overexpression of USP44 contributes to the pathogenesis of T-ALL by regulating CDC20-APC/C activity and chromosome instability [8].
MicroRNAs (miRNAs) are a category of small noncoding RNAs play important roles in T-ALL [9]. For instance, the downregulation of miR-101 promotes the expression of TAL1, whose high expression induces stem-cell-like transcriptional circuitry in T-ALL [10]. Overexpression of miR-1246/1248 could regulate NOTCH2 pathway to promote cell proliferation in the T-ALL [11]. Moreover, TFs and miRNAs may co-regulate target genes to form feed-forward loops (FFLs), which serve as critical motifs in gene regulatory networks and play critical roles in multiple biological processes [12, 13]. The FFLs could reduce the complexity of regulatory networks and provide comprehensive clues to identify oncogenes and the underlying regulatory mechanisms [14]. BRCA1 and miR-25 targeting ADAMTSL3 led to better survival in colorectal cancer [15]. TF STAT5A and miR-146b-5p co-regulated the expression of NUMB to play important regulation on leukemogenesis by enhancing the ROS level and genome instability [16]. miR-19b represses CYLD expression and upregulates NFKB expression to active NF-KB pathway in T-ALL, and TCF3 regulates miR-125b and MYC in AML pathways [17, 18]. Thus, dissecting regulatory networks and exploring FFLs consisted of TF-miRNA-targets could provide profound insights to reveal the molecular pathogenesis of T-ALL.
In this study, we analyzed the alteration of transcriptional profiling including genes and miRNAs between T-ALL and normal T cells. Functional enrichment and TF-miRNA regulatory network analyses identified that FOXM1, MYB, SOX4 and miR-21/19b as core regulators to regulate the cell cycle related processes. Besides, CDC25A and CAPN2 may be potential targets for the treatment of T-ALL. This work will be helpful to enhance the understanding of pathogenesis as well as therapy for T-ALL.
Methods
Data sources and differential expression of miRNAs and genes
For the mRNA gene expression, we selected the GSE48558 dataset from GEO database (15 T-ALL cell lines, 13 T-ALL patient samples and 17 normal T cell samples), in which T-ALL cell lines included CEM, JURKAT, MOLT and KARPAS45 [19]. GEO2R [20] was used to compare the gene expressions of T-ALL cell lines and T-ALL patient samples with normal T cells, respectively. Benjamini & Hochberg method was used to adjust the p-value, and p-value <1e-5 and fold change (FC) > 4 were considered as significant difference. The intersection of these two comparisons was considered as differentially expressed genes (DEGs) of T-ALL and normal samples. Then, the DAVID Tool [21] was applied to perform enrichment analysis for upregulated and downregulated DEGs, respectively.
miRNA datasets were obtained from GSE89978 (48 T-ALL patient samples, 2 CD4+ CD8+ and 2 CD34+ healthy donor samples) [22]. In these datasets, T-ALL lymphoblasts collected from the blood samples and bone marrow of T-ALL patients, and CD4+ CD8+/CD34+ cells were obtained from healthy donor thymocytes. Using DESeq2, we compared the T-ALL samples with CD4+ CD8+ and CD34+ normal samples, respectively. We required FC > 2, p-value < 0.05 and read count > 100 in at least 1 sample as the cutoffs for differential expression. To avoid the bias caused by the unbalanced sample size in the different groups, RNentropy was employed to evaluate the robust of miRNAs expression, and the miRNAs with convergent expression profiles were used for further analyses [23]. Finally, we selected miRNAs overlapped in two comparisons as differentially expressed miRNAs (DEMs). The MeV (https://sourceforge.net/projects/mev-tm4/files/) software with hierarchical clustering method was used to present the expression heatmap by showing the mean expression.
Generation of network and analysis of hub components
Briefly, the miRNA-TF-target regulatory network was constructed by the following three components: 1) TF-targets regulation. TF-targets regulatory information was obtained from the ChIP-Seq experimental datasets of public databases (ENCODE, and hTFtarget http://bioinfo.life.hust.edu.cn/hTFtarget/) and predictive TF-targets from AnimalTFDB [24] and UCSC; 2) miRNA-targets regulation. Experimentally validated miRNA-targets regulations were incubated from miRTarBase and TarBasev7.0, while the predictive regulations were collected form Targetscan and miRanda; 3) Regulatory network detection. The detailed procedures about how to select nodes to form potential FFLs were described in our previous studies [14, 25]. In this study, the DEMs and DEGs were used to build the regulatory network, and all networks were visualized by Cytoscape (version 3.4.0).
NetworkAnalyzer was employed to calculate the degree of nodes in the network and helped to identify the hubs. The subnetwork was built by the DEGs enriched in the GO terms and the corresponding regulatory DEMs and TFs. Based on the subnetwork, the top 5 TFs and miRNAs that contained the maximum connection combined with their targets were extracted to find the core TFs and miRNAs. The half maximal inhibitory concentration (IC50) information of 746 drugs and the corresponding genes expression profiles of 1861 cell lines were collected from GDSC [26] and CTRP [27] databases. The spearman correlation coefficient (cor_sprm) between the IC50 and genes expression profiles were calculated in all cell lines using the GSCALite [28], and the drug-gene pair with the p-value <1e-4 & |cor_sprm| > 0.4 was considered as significant one for the further analysis.
Results
Transcriptional profiling analysis reveals significant upregulation of cell proliferation and downregulation of immune response in T-ALL
To explore the alterations of transcriptional profiling involved in the progress of T-ALL, we detected the DEGs and DEMs by comparing T-ALL samples with normal samples. As a result, we identified 1141 and 604 DEGs in the comparisons of T-ALL cell lines vs normal T-cells and T-ALL patients vs normal T cells, respectively. There were 434 DEGs overlapped with the same trends (244 upregulated and 190 downregulated) in the two comparisons (Fig. 1b). Additionally, we found 24 DEMs in the comparison of T-ALL samples vs normal T cells (Fig. 2a). Among them, 20 miRNAs were upregulated and 4 miRNAs were downregulated in T-ALL.

Differential expression analysis in T-ALL vs normal T-cells. (a) GO enrichment results of upregulated DEGs (red). (b) Venn graph of DEGs in the two comparisons. C: T-ALL Cell lines, N: Normal human T-cells, P: T-ALL patients. Numbers in the sectors are the numbers of DEGs downregulated (green) and upregulated (red). (c) GO enrichment results of downregulated DEGs (green)

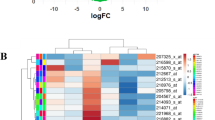
(a) Hierarchical clustering of miRNAs significantly differentially expressed in the comparisons of T-ALL samples (patients and cell lines) vs normal T cells (b) Hierarchical clustering of TFs. Upregulation in red and downregulation in green
For further investigating the functions of DEGs in T-ALL, we performed functional enrichment analysis on the DEGs. Interesting, more than 50% (130/244) upregulated DEGs were enriched in the processes related to cell cycle, such as cell division, cell proliferation and DNA replication (Fig. 1a). Uncontrolled cell cycle and cell proliferation led to the abnormal cell growth of T-cells [29], which suggested that these genes played important roles in the development of T-ALL. The downregulated genes were enriched in immune related processes and cell surface receptor signaling (Fig. 1c), which implied that the immune system of T-ALL patients was suppressed.
Regulatory network involved in the development of T-ALL
TFs are key regulators of gene expression and play vital roles in the pathogenesis of T-ALL. Among the 434 DEGs, 31 differentially expressed TFs (13 up and 18 down) were identified and their expression hierarchical clustering analysis was showed in Fig. 2b. For example, the downregulation of MYB and SOX4 could suppress the migration, cell proliferation and development of T-ALL [30, 31]. Meanwhile, downregulation of JUNB by miR-149 promoted T cell proliferation and suppressed apoptosis [32].
For understanding the regulation relationships among TFs, miRNAs and their targets, we constructed the regulatory network based on the DEGs and DEMs. Our network contained 486 edges that consisted of 132 DEGs (14 TFs and 118 genes) and 12 miRNAs (Fig. 3). Meanwhile, NR4A2, MYB, SOX4 were the top 3 TFs in the connected degree, and FOXM1 was the only TF enriched in cell cycle related pathways, while FOXM1, MYB and SOX4 regulated about the amount of 66% of the genes and all the miRNAs in the network. miR-21-5p, miR-19b-3p and miR-132-3p were the top 3 miRNAs which regulated about 66% of the genes in our network. In conjunction with the results above, the hub TFs and miRNAs combined with their target genes in our regulatory network may form key modules involved in the development of T-ALL.

The regulatory network of DEGs and DEMs. Green, downregulated genes and miRNAs. Red, upregulated genes and miRNAs. The diamond nodes, TFs; Ellipse nodes, DEMs; Round Rectangle, DEGs. The size of the nodes represents the degree of the nodes
Key regulatory modules participate in the progression of T-ALL
To reveal how the hub regulators affect cell proliferation and result in T-ALL, we reconstructed a subnetwork using hub genes which enriched in cell cycle/proliferation processes and were with high connection property to TFs and miRNAs (Fig. 4). Totally, 51 nodes with 69 FFLs were contained in the subnetwork. We detected 3 hub TFs (FOXM1, MYB and SOX4), which cooperated with 11 miRNAs to regulate most genes (18/25) in these subnetwork.

TF and miRNA regulatory subnetwork of cell cycle and cell proliferation genes. Light blue Round Rectangle, enriched GO terms; Grey line, regulatory relationship of TFs and miRNAs to genes; Blue line, relationship of mRNAs to GO terms. The means of nodes are the same as Fig. 3
To further explore the functions of key regulators, we dissected the subnetworks and extracted important FFLs which may play vital roles underlying the development of T-ALL (Fig. 5). The FFLs FOXM1-miR-21-5p-CDC25A and SOX4-miR-19b-3p-RBBP8 were predicted to regulate cell cycle and cell division. FOXM1 could regulate the expression of target gene CDC25A, which was considered as a symbol to characterize the accumulation of G1/S and early S phase cells and the downregulation of CDC25A induced cell cycle arresting in T-ALL [33]. Meanwhile, inhibition the expression of FOXM1 could decrease the proliferation of Jurkat cells and improved the survival of children T-ALL [34]. SOX4/MYB could regulate the expression of RBBP8, which play important roles in DNA repair [35].

FFLs for FOXM1-miR-21-CDC25A and MYB/SOX4-miR-19b-RBBP8 and their enriched GO terms. The means of nodes are the same as Fig. 3
To investigate the possible functions of miR-21-5p and miR-19b-3p in the FFL and the development of T-ALL, we analyzed the target genes of the two miRNAs using DAVID. Interestingly, the functions of targets genes for miR-21-5p were focused on regulation of apoptosis and cell death processes (Fig. 6a), which demonstrated that miR-21-5p played important roles in cell cycle regulation. Interestingly, miR-21-5p was observed overexpressed in both early and late-stage primary murine T-ALL cells, while knocked down of miR-21-5p induced T-ALL cell apoptosis [36]. Furthermore, miR-21-5p could regulate CDC25A participating the cell cycle process [37]. Meanwhile, the targets of miR-19b were mainly related to regulation of transcription (Fig. 6b), and miR-19b could induce leukemogenesis under NOTCH1 overexpression [38]. Thus, the above results illustrated, FOXM1-miR-21-5p-CDC25A and SOX4-miR-19b-3p-RBBP8 may serve as core regulatory modules involved in the development of T-ALL.

GO terms enrichments result for the target genes of miR-21-5p (a) and miR-19b-3p (b)
Potential drug indicators of genes in the regulatory network
For identifying the potential therapeutic indication of the regulatory network, we analyzed the correlation between IC50 of drugs and regulators in our network, and constructed the detail a drug-FFL regulatory network (Fig. 7). Totally, 34 genes were highly correlated with 72 drugs or small molecules, ANLN, CAPN2, C4orf46, SYTL2, CDC25A, MCM2 and KLRG1 were the most significant genes with highest correlation to more than 10 drugs. GSK-J4, BRD-K30748066, tozasertib and Teniposide were the top 4 drugs correlated with many genes. GSK-J4 as small molecule inhibitor could suppress the growth of T-ALL cells, and its target KDM6A/UTX acted as a pro-oncogenic cofactor in T-ALL [39]. Teniposide was reported in several drug combinations in treatment of T-ALL, showed ability to induce cell death [40]. These results implied that these genes may be efficient biomarkers and these drugs may be able to target these genes to treat T-ALL.

The correlation of drugs IC50 and genes in the regulatory network of FOXM1, MYB, SOX4, miR-21-5p and miR-19b-3p. Drug names are in red font. Upregulation significance genes are red; downregulation significance genes are green. Orange or purple dots mean positive or negative correlation between drugs IC50 and genes expression, respectively
Besides, CDC25A, which identified as a core target gene of FOXM1-miR-21-5p-CDC25A, showed negative correlation with drug which inhibited T-ALL, such as GSK-J4 and Teniposide. Meanwhile, CAPN2 was downregulated in T-ALL sample and positive correlated with 41 drugs. Beta carotene can increase the expression of BAX and CAPN2 and lead to the apoptosis of AML cell lines [41]. Furthermore, abnormally high expression of MCM2 was reported in recurrence of ALL [42], we found MCM2 was upregulated in the T-ALL samples and showed negative correlation with 39 drugs, which implied MCM2 may serve as a potential therapeutic target. Thus, our regulatory network may serve as a resource to explore the development of T-ALL and provide potential targets for drug treatment.
Discussion
The T-ALL still remains a cancer with worse prognosis for high recurrence, understanding the regulatory mechanisms of T-ALL could provide comprehensive insights for the diagnosis and treatment. In current study, we investigated the potential regulatory mechanisms of the development of T-ALL through differential expression and network analysis across T-cells, T-ALL and normal samples. Our data demonstrated that the biological processes including cell cycle, DNA replication and cell proliferation were enhanced, but immune response was decreased in T-ALL. We found that FOXM1, MYB, SOX4 and miR-21/19b as hub regulators may form FFLs with CDC25A and RBBP8 to regulate cell cycle related pathways. Drug targets analysis could provide preliminary clues that CAPN2, CDC25A and MCM2 may serve as potential therapeutic targets for the treatment of T-ALL.
Understanding the regulatory mechanisms underlying the development of T-ALL could provide profound insights to reveal the molecular pathogenesis of T-ALL. Most of the previous studies were focused on the effects of single gene or pathways in T-ALL, rare reports were involved in the regulatory relationships on systems level. Our results demonstrated that genes involved in the processes of cell proliferation, cell cycle and DNA replication were upregulated in the T-ALL samples (Fig. 1), which indicated the unexpected cell proliferation occurred in the T-ALL. Several researchers reported that cell proliferation, cell cycle and DNA replication pathways played crucial roles in T-ALL, and blocked down genes related to cell cycle could contribute to the remission of T-ALL. For example, resveratrol inhibited Akt/mTOR and activated p38-MAPK pathways could induce apoptosis of malignant cells in T-ALL [43]. Dysregulation for the genes involved in the DNA replication processes could cause differentiation blockade of normal T-cells, which in turn results in the initiation of T-ALL [44]. Our regulatory analysis showed that the TFs FOXM1, MYB, SOX4 and miR-21/19b represented as hub nodes and core regulators in the T-ALL regulatory network (Fig. 4). In our key regulatory networks, the TF FOXM1 and miR-21 predicted co-regulating target gene CDC25A, and formed a FFL participating the DNA replication, cell proliferation and division processes (Fig. 5), which may be a key module involved in the development of T-ALL. The TF FOXM1 was reported as a core factor involved in the cellular transformation and tumor initiation by regulating the G1/S and G2/M transitions [45], while the miR-21 could modulate cell proliferation and migration [46]. Meanwhile, downregulation of CDC25A was concordant with the inhibition of Jurkat cell proliferation in a G2/M arrest [33]. Furthermore, the expression profiles of the FOXM1-miR-21-5p-CDC25A FFL loop was consistent with the above reports, which implied the vital regulatory roles of the FFL in the development of T-ALL. The upregulated expression of the TF FOXM1 could increase the expression of both CDC25A [47] and miR-21-5p, whereas the miR-21 could target CDC25A [48], which in turn formed a dynamic balance for the FFL involved in the cell proliferation process, suggesting the miR-21 may mediate a negative feedback loop collaborated with the TF FOXM1 to regulate their targets CDC25A. Consequently, although the miR-21 repressed the expression of CDC25A, the CDC25A maintained at a high expression level promoted by TF FOXM1, which may trigger the abnormal cell cycle and cell proliferation in the progress of T-ALL. Additionally, SOX-miR-19b-3p-RBBP8 may reveal the regulatory relationships involved in the T-ALL as well. SOX4 could enhance chondrogenic differentiation and proliferation of human synovium-derived stem cell [49], while downregulation of SOX4 suppressed cell proliferation and induced apoptosis [50], which may contribute to remission of T-ALL. miR-19b was reported regulating cell cycle related pathways, and inhibited the activity of the PI3K-AKt signaling pathway leading to inactivation of P53 and cell growth in cancers [51]. RBBP8, also called CtIP, promoted DNA-end resection and maintained chromosome stability [35], and depletion of CtIP leading to cell arrested in G1 phase [52]. In conjunction of above results, FOXM1-miR-21-5p-CDC25A and SOX-miR-19b-3p-RBBP8 FFL may regulate cell cycle related processes playing profound roles in the development of T-ALL.
Regulatory network combined with drug-specific analyses employed the gene expression, gene regulatory relationships and IC50 of drug to seek potential drug indicators involved in the development of T-ALL. Our data demonstrated that GSK-J4 showed high correlations with most genes in the regulatory network (Fig. 7), which implied that GSK-J4 may serve as a potential effective drug for T-ALL. Interesting, previous studies reported that GSK-J4 could decrease the activity of H3K27 demethylases JMJD3, which may inhibit NOTCH1 related pathways and contribute to the anticarcinogen effects [53]. Meanwhile, gene EZH2, which was associated with H3K27 methylation and activated in dividing cells [54], showed highest negative correlation with GSK-J4. EZH2 was highly expressed in several human tumors, whose upregulation promoted cell proliferation and amplified in cancer [55]. Additionally, increase the activity of CAPN2 could eliminate malignant cells in ATL cells, and decrease of CAPN2 expression was observed in ATL patients [56]. FOXM1 and miR-21-5p co-regulated the expression of CAPN2 in our FFL, which may be involved in the T-ALL. The expression level of CAPN2 was downregulated in the T-ALL patients, and displayed high positive correlation with the drug GSK-J4, which suggested CAPN2 may be acted as potential drug target in the T-ALL. Combined with above results, our drug indicators analysis may provide some potential drugs and targeted biomarker in the therapy of T-ALL.
Conclusions
We analyzed the alteration of transcriptome profiling and regulatory networks between T-ALL sample and normal T cell samples at transcriptional and post-transcriptional levels. Our research identified some dysregulated genes enriched in cell cycle related processes. TF-miRNA co-regulatory network analysis demonstrated that FOXM1, MYB, SOX4, miR-21/19b and their FFLs FOXM1-miR-21-5p-CDC25A and MYB/SOX4-miR-19b-3p-RBBP8 may play important roles in the cell proliferation of T-ALL. Consequently, our study revealed the regulatory relationships in the cell cycle related processes of T-ALL and may provide potential therapeutic targets for T-ALL.
Abbreviations
- Cor_sprm:
-
Spearman correlation coefficient
- DEG:
-
Differentially expressed gene
- DEM:
-
Differentially expressed miRNA
- FC:
-
Fold change
- FFL:
-
Feed-forward loop
- IC50:
-
Half maximal inhibitory concentration
- miRNA:
-
microRNA
- T-ALL:
-
T-cell acute lymphoblastic leukemia
- TF:
-
Transcription factor
References
Liu X, Liu S, Chen J, He L, Meng X, Liu S. Baicalein suppresses the proliferation of acute T-lymphoblastic leukemia Jurkat cells by inhibiting the Wnt/β-catenin signaling. Ann Hematol. 2016;95(11):1787–93.
Sanda T, Leong WZ. TAL1 as a master oncogenic transcription factor in T-cell acute lymphoblastic leukemia. Exp Hematol. 2017;53:7–15.
Durinck K, Goossens S, Peirs S, Wallaert A, Van Loocke W, Matthijssens F, et al. Novel biological insights in T-cell acute lymphoblastic leukemia. Exp Hematol. 2015;43(8):625–39.
Blank U, Karlsson G, Karlsson S. Signaling pathways governing stem-cell fate. Blood. 2008;111(2):492–503.
Collins C, Wang J, Miao H, Bronstein J, Nawer H, Xu T, et al. C/EBPα is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis. Proc Natl Acad Sci U S A. 2014;111(27):9899–904.
Neumann M, Vosberg S, Schlee C, Heesch S, Schwartz S, Gökbuget N, et al. Mutational spectrum of adult T-ALL. Oncotarget. 2014;6(5):2754–66.
Sarmento LM, Póvoa V, Nascimento R, Real G, Antunes I, Martins LR, et al. CHK1 overexpression in T-cell acute lymphoblastic leukemia is essential for proliferation and survival by preventing excessive replication stress. Oncogene. 2015;34(23):2978–90.
Zhang Y, van Deursen J, Galardy PJ. Overexpression of ubiquitin specific protease 44 (USP44) induces chromosomal instability and is frequently observed in human T-cell leukemia. PLoS One. 2011;6(8):e23389.
Ma Z, Liu T, Huang W, Liu H, Zhang H-M, Li Q, et al. MicroRNA regulatory pathway analysis identifies miR-142-5p as a negative regulator of TGF-β pathway via targeting SMAD3. Oncotarget. 2016;7(44):71504–13.
Correia NC, Melão A, Póvoa V, Sarmento L, Gómez de Cedrón M, Malumbres M, et al. microRNAs regulate TAL1 expression in T-cell acute lymphoblastic leukemia. Oncotarget. 2016;7(7):8268–81.
Luo M, Zhang Q, Xia M, Hu F, Ma Z, Chen Z, et al. Differential co-expression and regulatory network analysis uncover the relapse factor and mechanism of T cell acute leukemia. Molecular Therapy - Nucleic Acids. 2018;12:184–94.
Siciliano V, Garzilli I, Fracassi C, Criscuolo S, Ventre S, di Bernardo D. miRNAs confer phenotypic robustness to gene networks by suppressing biological noise. Nat Commun. 2013;4.
Lin Y, Zhang Q, Zhang H-M, Liu W, Liu C-J, Li Q, et al. Transcription factor and miRNA co-regulatory network reveals shared and specific regulators in the development of B cell and T cell. Sci Rep. 2015;5.
Zhang H-M, Kuang S, Xiong X, Gao T, Liu C, Guo A-Y. Transcription factor and microRNA co-regulatory loops: important regulatory motifs in biological processes and diseases. Brief Bioinform. 2015;16(1):45–58.
Wang H, Luo J, Liu C, Niu H, Wang J, Liu Q, et al. Investigating MicroRNA and transcription factor co-regulatory networks in colorectal cancer. BMC bioinformatics. 2017;18(1):388.
Zhang H-M, Li Q, Zhu X, Liu W, Hu H, Liu T, et al. miR-146b-5p within BCR-ABL1-positive microvesicles promotes leukemic transformation of hematopoietic cells. Cancer Res. 2016;76(10):2901–11.
Lin XC, Xu Y, Sun GP, Wen JL, Li N, Zhang YM, et al. Molecular dysfunctions in acute myeloid leukemia revealed by integrated analysis of microRNA and transcription factor. Int J Oncol. 2016;48(6):2367–80.
Ye H, Liu X, Lv M, Wu Y, Kuang S, Gong J, et al. MicroRNA and transcription factor co-regulatory network analysis reveals miR-19 inhibits CYLD in T-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2012;40(12):5201–14.
Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, Padget M, Irvine DA, Sliwinski T, et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. 2013;122(7):1293–304.
Davis S, Meltzer PS. GEOquery: a bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics (Oxford. England. 2007;23(14):1846–7.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Wallaert A, Van Loocke W, Hernandez L, Taghon T, Speleman F, Van Vlierberghe P. Comprehensive miRNA expression profiling in human T-cell acute lymphoblastic leukemia by small RNA-sequencing. Sci Rep. 2017;7(1):7901.
Zambelli F, Mastropasqua F, Picardi E, D’Erchia AM, Pesole G, Pavesi G. RNentropy: an entropy-based tool for the detection of significant variation of gene expression across multiple RNA-Seq experiments. Nucleic Acids Res. 2018;46(8):e46.
Hu H, Miao YR, Jia LH, Yu QY, Zhang Q, Guo AY. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2018.
Lin Y, Sibanda VL, Zhang H-M, Hu H, Liu H, Guo A-Y. MiRNA and TF co-regulatory network analysis for the pathology and recurrence of myocardial infarction. Sci Rep. 2015;5:9653.
Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of drug sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41(Database issue):D955–61.
Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154(5):1151–61.
Liu CJ, Hu FF, Xia MX, Han L, Zhang Q, Guo AY. GSCALite: a web server for gene set cancer analysis. Bioinformatics. 2018;34(21):3771–2.
Rahmati M, Moosavi MA, Zarghami N. Nucleostemin knocking-down causes cell cycle arrest and apoptosis in human T-cell acute lymphoblastic leukemia MOLT-4 cells via p53 and p21Waf1/Cip1 up-regulation. Hematology (Amsterdam, Netherlands). 2014;19(8):455–62.
Mets E, Van der Meulen J, Van Peer G, Boice M, Mestdagh P, Van de Walle I, et al. MicroRNA-193b-3p acts as a tumor suppressor by targeting the MYB oncogene in T-cell acute lymphoblastic leukemia. Leukemia. 2015;29(4):798–806.
Yin J-J, Liang B, Zhan X-R. MicroRNA-204 inhibits cell proliferation in T-cell acute lymphoblastic leukemia by down-regulating SOX4. Int J Clin Exp Pathol. 2015;8(8):9189–95.
Fan SJ, Li HB, Cui G, Kong XL, Sun LL, Zhao YQ, et al. miRNA-149* promotes cell proliferation and suppresses apoptosis by mediating JunB in T-cell acute lymphoblastic leukemia. Leuk Res. 2016;41:62–70.
Jazi MS, Mohammadi S, Yazdani Y, Sedighi S, Memarian A, Aghaei M. Effects of valproic acid and pioglitazone on cell cycle progression and proliferation of T-cell acute lymphoblastic leukemia Jurkat cells. Iranian Journal of Basic Medical Sciences. 2016;19(7):779–86.
Wang J-Y, Jia X-H, Xing H-Y, Li Y-J, Fan W-W, Li N, et al. Inhibition of Forkhead box protein M1 by thiostrepton increases chemosensitivity to doxorubicin in T-cell acute lymphoblastic leukemia. Mol Med Rep. 2015;12(1):1457–64.
Hühn D, Kousholt AN, Sørensen CS, Sartori AA. miR-19, a component of the oncogenic miR-17∼92 cluster, targets the DNA-end resection factor CtIP. Oncogene. 2015;34(30):3977–84.
Junker F, Chabloz A, Koch U, Radtke F. Dicer1 imparts essential survival cues in notch-driven T-ALL via miR-21-mediated tumor suppressor Pdcd4 repression. Blood. 2015;126(8):993–1004.
Hu B, Wang X, Hu S, Ying X, Wang P, Zhang X, et al. miR-21-mediated Radioresistance occurs via promoting repair of DNA double Strand breaks. J Biol Chem. 2017;292(8):3531–40.
Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol. 2010;12(4):372–9.
Benyoucef A, Palii CG, Wang C, Porter CJ, Chu A, Dai F, et al. UTX inhibition as selective epigenetic therapy against TAL1-driven T-cell acute lymphoblastic leukemia. Genes Dev. 2016;30(5):508–21.
Winter SS, Holdsworth MT, Devidas M, Raisch DW, Chauvenet A, Ravindranath Y, et al. Antimetabolite-based therapy in childhood T-cell acute lymphoblastic leukemia: a report of POG study 9296. Pediatr Blood Cancer. 2006;46(2):179–86.
Sacha T, Zawada M, Dulińska-Litewka J, Lach Z, Szostek M, Bodzioch M, et al. Beta-carotene regulates the expression of proapoptotic BAX and CAPN2 in HL-60, U-937 and TF-1 - human acute myeloid leukemia cell lines; microarray, RQ-PCR and Western blot analysis. Przeglad Lekarski. 2011;68(5):258–62.
Li S, Wang C, Wang W, Liu W, Zhang G. Abnormally high expression of POLD1, MCM2, and PLK4 promotes relapse of acute lymphoblastic leukemia. Medicine. 2018;97(20):e10734.
Ge J, Liu Y, Li Q, Guo X, Gu L, Ma ZG, et al. Resveratrol induces apoptosis and autophagy in T-cell acute lymphoblastic leukemia cells by inhibiting Akt/mTOR and activating p38-MAPK. Biomedical and environmental sciences: BES. 2013;26(11):902–11.
Sincennes M-C, Humbert M, Grondin B, Lisi V, Veiga DFT, Haman A, et al. The LMO2 oncogene regulates DNA replication in hematopoietic cells. Proc Natl Acad Sci U S A. 2016;113(5):1393–8.
Koo C-Y, Muir KW, Lam EW-F. FOXM1: from cancer initiation to progression and treatment. Biochim Biophys Acta. 2012;1819(1):28–37.
Xu J, Zhang W, Lv Q, Zhu D. Overexpression of miR-21 promotes the proliferation and migration of cervical cancer cells via the inhibition of PTEN. Oncol Rep. 2015;33(6):3108–16.
Sullivan C, Liu Y, Shen J, Curtis A, Newman C, Hock JM, et al. Novel interactions between FOXM1 and CDC25A regulate the cell cycle. PLoS One. 2012;7(12):e51277.
Wang P, Zou F, Zhang X, Li H, Dulak A, Tomko RJ, et al. microRNA-21 negatively regulates Cdc25A and cell cycle progression in colon cancer cells. Cancer Res. 2009;69(20):8157–65.
Zhang L, Chen S, Bao N, Yang C, Ti Y, Zhou L, et al. Sox4 enhances chondrogenic differentiation and proliferation of human synovium-derived stem cell via activation of long noncoding RNA DANCR. J Mol Histol. 2015;46(6):467–73.
Zhou Y, Wang X, Huang Y, Chen Y, Zhao G, Yao Q, et al. Down-regulated SOX4 expression suppresses cell proliferation, metastasis and induces apoptosis in Xuanwei female lung cancer patients. J Cell Biochem. 2015;116(6):1007–18.
Farooqi AA, Yaylim I, Ozkan NE, Zaman F, Halim TA, Chang H-W. Restoring TRAIL mediated signaling in ovarian cancer cells. Arch Immunol Ther Exp. 2014;62(6):459–74.
Chen P-L, Liu F, Cai S, Lin X, Li A, Chen Y, et al. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol Cell Biol. 2005;25(9):3535–42.
Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature. 2014;514(7523):513–7.
Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–9.
Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 2003;22(20):5323–5335.
Ishihara M, Araya N, Sato T, Tatsuguchi A, Saichi N, Utsunomiya A, et al. Preapoptotic protease calpain-2 is frequently suppressed in adult T-cell leukemia. Blood. 2013;121(21):4340–7.
Acknowledgements
We are grateful to the groups submitted the dataset GSE48558 and GSE89978 to GEO database.
Availability of data and material
The data used in this study are from public database [NCBI GEO: GSE48558 and GSE89978].
Funding
This study was supported by National Natural Science Foundation of China (NSFC) [31822030, 31801113 and 31801154], China Postdoctoral Science Foundation [2017 M622455 and 2018 M632830], Key Research & Development Plan of Shandong Province [2015GSF118031], and program for HUST Academic Frontier Youth Team.
Author information
Authors and Affiliations
Contributions
QZ, QL, AYG designed the study. PL, YW collected and prepared the data. MX, ML performed analyses. MX drafted the manuscript. MX, QZ, QL, AYG revised manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xia, M., Zhang, Q., Luo, M. et al. Regulatory network analysis reveals the oncogenesis roles of feed-forward loops and therapeutic target in T-cell acute lymphoblastic leukemia. BMC Med Genomics 12, 8 (2019). https://doi.org/10.1186/s12920-018-0469-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-018-0469-0