
Abstract
Background
The PALB2 gene encodes a protein that plays a crucial role in maintaining genomic integrity. Germline inactivating mutations in PALB2 are associated with an increased risk of breast and ovarian cancer. The prevalence and spectrum of recurrent PALB2 germline mutations in breast and ovarian cancer patients from Poland is not clearly defined.
Methods
PALB2 exons were amplified from 460 BRCA1/2-mutation negative women with familial breast and/or ovarian cancer and early-onset breast cancer using AmpliSeq technology and sequenced on an Ion Torrent PGM sequencer. In addition, eight selected variants were genotyped using TaqMan assays in 807 BRCA1/2-mutation negative breast cancer patients and 1690 healthy women.
Results
Two recurrent PALB2 mutations, c.172_175delTTGT and c.509_510delGA, were identified, along with one novel mutation, c.347insT. In total, PALB2 pathogenic mutations were detected in 7/460 (1.5%) patients. Furthermore, in breast and/or ovarian cancer patients, several single nucleotide variants (SNVs) were detected in the PALB2 coding region. In an additional group of 807 patients, eight (1%) carriers of two pathogenic mutations, c.172_175delTTGT (0.5%) and c.509_510delGA (0.5%), were identified. The c.509_510delGA mutation was not identified in healthy controls, while c.172_175delTTGT was identified in 4/1690 (0.24%) of control women.
Conclusions
Germline mutations in the PALB2 gene were observed at a frequency of approximately 1.5% in Polish breast and/or ovarian cancer patients. Our study confirms two recurrent PALB2 mutations; c.172_175delGA and c.509_510delGA.
Similar content being viewed by others


Background
Breast cancer is the most frequently diagnosed malignancy and the leading cause of cancer death in women worldwide [1]. About 5–10% of breast and 10% of ovarian cancers are thought to be hereditary [2]. Mutations in the two main susceptibility genes, BRCA1 and BRCA2, account for 20% of hereditary breast cancer (HBC) and 30% of hereditary breast and ovarian cancer (HBOC) [3]. Other genes participating in DNA-damage response pathways, including CHEK2, NBS1, ATM, BRIP1, and PALB2, are also involved in HBC and HBOC [4, 5].
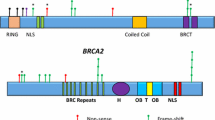
The PALB2 (MIM 610355, Partner and Localizer of B RCA 2) gene was identified in a search for novel components of endogenous BRCA2 containing complexes [6]. The PALB2 protein interacts with both BRCA1 and BRCA2 through its N-terminal coiled-coil and C-terminal WD-40 domains, respectively. These three proteins form a “BRCA complex” in which PALB2 acts as a bridge between BRCA1 and BRCA2. The “BRCA complex” is crucial for initiating homologous recombination in the DNA-damage response [6–8].
Biallelic inactivating germline mutations in the PALB2 gene lead to Fanconi anemia (Fanconi anemia type N), whereas monoallelic mutations are associated with an increased risk of breast, pancreatic, and possibly ovarian cancer [9–14]. Germline mutations in PALB2 are responsible for 1–3.9% of HBC [15–26]. Similar to BRCA2, germline mutations in PALB2 are also associated with hereditary predisposition to male breast cancer. To date, approximately 50 truncating mutations in PALB2 have been detected in breast cancer families worldwide.
The first breast cancer family-based association study estimated that a relative risk of 2.3 (95% CI: 1.4–3.9) is conferred by mutations of PALB2 [12]. Most recently, Antoniou and colleagues. applied a modified segregation procedure to show that the age-specific risk of breast cancer in female mutation carriers overlaps with that conferred by BRCA2 mutations [27].
In a previous study, we demonstrated that in a group of 512 patients for whom targeted BRCA1/2 mutation testing did not show any pathogenic variants, sequencing of BRCA1/2 exons identified 52 carriers of mutations [28, 29]. The aim of the present study was to investigate the contribution of PALB2 germline mutations in a group of 460 BRCA1/2-mutation negative breast and/or ovarian cancer patients and identify the optimal panel of recurrent mutations for genetic screening of PALB2.
Methods
Patients
DNA samples were chosen from the repository of the Genetic Counseling Unit, Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology in Warsaw. In-depth interviews assessed personal and familial cancer history. Healthy women with no known history of cancer were enrolled from the National Colorectal Cancer Screening Program. Patients comprised a group of 460 BRCA1/2-mutation negative breast cancer patients (with a median age of 43 years, range 17–68 years) and included 165, 103, and 192 women with HBC, HBOC, and early-onset breast cancer (EOBC), respectively [29]. This group of patients had undergone full BRCA1/2 sequencing using the Ion AmpliSeq BRCA1 and BRCA2 Panel (Thermo) on a PGM sequencer as part of our previous study [29].
An additional group, comprising 807 selected BRCA1/2-mutation negative women, was surveyed using TaqMan SNP genotyping assays. This group had a median age of 48 years (range 20–85 years); included 322, 81, and 404 HBC, HBOC, and EOBC patients, respectively; and were negative for 20 selected BRCA1/2 mutations, including 11 mutations in BRCA1, namely, c.66_67delAG, c.181T > C, c.3756delGTCT, c.3700_3704del5, c.4035delA, c.3777delT, c.4065delTCAA, c.4041delAG, c.5263delC, c.5213G > A, and 5370C > T, and nine mutations in BRCA2, namely, c.1408G > T, c.5946delT, c.5239insT, c.6447delTA, c.5964delAT, c.7910del5, c.9382C > T, c.8924delT, and c.9402delT [28].
NGS of PALB2
Genomic DNA was extracted from peripheral blood, as previously described [28]. DNA concentrations were determined using a Qubit 2.0 Fluorometer (Thermo) and the dsDNA HS Assay Kit (Thermo). For library construction, the Ion AmpliSeq™ Library Kit 2.0 (Thermo) and Ion AmpliSeq PALB2 custom primers, comprising 27 primer pairs spanning 5.29 kb, were used to amplify the coding regions of the PALB2 gene. Library concentrations and size distributions were determined with the Qubit dsDNA HS Assay Kit (Thermo) and a High Sensitivity DNA Analysis Kit on a Bioanalyzer 2100 (Agilent), respectively.
Up to 48 libraries were combined in equimolar concentrations following their loading onto an Ion Chef for DNA template preparation with a set of reagents from an Ion PGM IC 200 Kit and an Ion 316 Chip Kit v2 BC (Thermo). Sequencing was performed on the Ion Torrent PGM in a mode with 500-flow runs. Data were analyzed on the Ion Torrent server using the Torrent Suite software (Thermo). Sequencing reads were aligned to the hg19 reference genome and fitted to PALB2 designed amplicons. The coverage analysis and variant caller plug-ins were run with optimal settings to identify germline variants. PALB2 gene variants were annotated using data from ClinVar-NCBI and LOVD.
TaqMan genotyping
Mutations found with NGS were validated by Sanger sequencing. Furthermore, eight selected variants, namely 172_175del4, c.347insT, c.509_510delGA, Gln559Arg, Glu672Gln, Leu939Trp, Gly998Glu, and Thr1100Thr were analyzed using TaqMan SNP Genotyping Assays (Thermo) on an ABI 7900HT qPCR system (Thermo), as previously described [28].
Results
NGS analysis
We evaluated the frequencies of PALB2 germline mutations in 460 BRCA1/2-mutation negative breast and/or ovarian cancer patients. Of the 460 women, 268 had HBC and/or HBOC, and 192 had EOBC, with a median age at breast cancer diagnosis of 43 years (range 17–68 years).
In this group, we identified seven unrelated carriers (1.5%) of the three PALB2 truncating mutations: c.172_175delTTGT (n = 4), c.347insT (n = 1), and c.509_510delGA (n = 2) (Table 1). Six of the seven carriers (2.2%) were among the 268 HBOC patients, and one (0.5%) was in the EOBC group.
In addition to these pathogenic mutations, we detected 10 SNVs in the coding region of the PALB2 gene (Table 2).
Genotyping of selected variants detected by NGS
Using TaqMan SNP genotyping assays, the frequencies of eight selected variants, including three truncating mutations (c.172_175delTTGT, c.347insT, c.509_510delGA), a rare missense variant (Leu939Trp), and four common polymorphisms (Gln559Arg, Glu672Gln, Gly998Glu, Thr1100Thr), were tested in an additional group of 807 breast and/or ovarian cancer patients (median age, 48 years; range 20–85 years), and 1690 healthy women (median age, 58 years; range 26–79 years).
The pathogenic mutation, c.172_175delTTGT, was identified in 4/807 (0.5%) breast cancer patients and in 4/1690 (0.24%) healthy controls (Table 2). The c.509_510delGA mutation was detected in four breast cancer patients, and was not found in healthy control subjects. The c.347insT mutation was identified in neither additional cancer patients nor healthy controls.
The frequencies of the four common polymorphisms were similar in cancer patients and healthy controls; therefore, the clinical significance of these polymorphic variants could not be confirmed. In addition, the Leu939Trp missense variant was detected in six of 807 (0.7%) cancer patients and three of 1690 (0.2%) healthy controls; hence, the incidence of this change was not significantly different between patients and controls.
Discussion
We identified three pathogenic PALB2 mutations among 460 BRCA1/2-mutation negative breast and/or ovarian cancer patients. The two recurrent mutations identified in our study were c.172_175delTTGT and c.509_510delGA. The novel pathogenic mutation c.347insT was found only in one young patient with HBC. The two recurrent mutations were identified in five patients with HBC and one patient with EOBC, but without any family history. In the cancer patient group, the recurrent variants (c.172_175delTTGT and c.509_510delGA) accounted for 86% (6/7) of PALB2 mutations. Our NGS and TaqMan genotyping studies indicate that the pathogenic c.509_510delGA mutation contributes to familial breast and/or ovarian cancer (0.9%); however, the c.172_175delTTGT mutation was identified both in familial (0.6%) and non-familial cancer patients (0.7%). The recurrent mutation c.509_510delGA is a European founder mutation that has been identified in breast cancer patients from Poland, Belarus, Russia, and Germany [16, 17, 24, 25]. This mutation has been detected in 0.6–1.4% of breast cancer patients in three independent studies in Poland [5, 16, 26]. The other recurrent PALB2 mutation, c.172_175delTTGT, was identified in breast cancer patients from the Czech Republic and Poland [5, 18, 26].
The PALB2 gene mutation rate (1.5%) determined for our group of breast and/or ovarian cancer patients was similar to those reported by other studies. PALB2 germline mutations have been identified in Finland, Germany, Poland, Czech Republic, Russia, Italy, Spain, Australia, United Kingdom, France, Netherlands, Canada, USA, China, and South Africa, with rates ranging from 0.1 to 3.9%, depending on the population [15–27]. In breast cancer families with both female and male breast cancer, PALB2 mutation rates increased to 6.7% in a UK study and to 9.0% in a Spanish study [12, 15].
In addition to clearly pathogenic mutations, we detected 10 missense variants in the coding region of the PALB2 gene. Identifying variants of unknown significance in cancer susceptibility genes represents a real problem in genetic counseling [30]. It should be stressed that, to date, no PALB2 missense variant has been classified as definitely pathogenic in any large population study [31]. Park et al. reported that the rare missense variant, Leu939Trp, is associated with altered direct binding of PALB2 to the RAD51C, RAD51, and BRCA2 proteins [32]. Functionally, the Leu939Trp mutant displays a decreased capacity for DNA double-strand break repair and an increased cellular sensitivity to ionizing radiation. In our group of breast and/or ovarian cancer patients, the missense variant, Leu939Trp, was detected in two (0.4%) breast cancer patients, one (0.4%) patient with HBOC, and in one (0.5%) with EOBC. Interestingly, in our study, the missense variant rate was 3.5 times more frequent in breast cancer patients (0.7%) than in healthy controls (0.2%), although the differences were not statistically significant. Consistent with this observation, a recent large case-control study by Southey et al. found no evidence of association of the Lys939Trp variant with breast cancer risk [33]. Furthermore, Catucci and colleagues [34] have also recently shown that the Lys939Trp variant does not disrupt the homologous recombination-mediated DNA repair activity of PALB2 and they concluded that this variant should be regarded as neutral with no clinical relevance to risk of breast cancer.
Conclusions
In summary, our study confirmed two PALB2 recurrent mutations, c.172_175delGA and c.509_510delGA, in BRCA1/2-mutation negative breast and/or ovarian cancer patients from Poland. The c.509_510delGA mutation contributed to familial cancer but was absent in non-familial cancer patients, while the c.172_175delTTGT mutation was identified in both familial and non-familial cancer patients.
Abbreviations
- BRCA1:
-
Breast cancer 1, early onset
- BRCA2:
-
Breast cancer 2, early onset
- EOBC:
-
Early-onset breast cancer
- HBC:
-
Hereditary breast cancer
- HBOC:
-
Hereditary breast and ovarian cancer
- NGS:
-
Next-generation sequencing
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012: Globocan 2012. Int J Cancer. 2015;136:E359–86.
Oosterwijk JC, de Vries J, Mourits MJ, de Bock GH. Genetic testing and familial implications in breast-ovarian cancer families. Maturitas. 2014;78:252–7.
Walsh T, King M-C. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–5.
Kuusisto KM, Bebel A, Vihinen M, Schleutker J, Sallinen S-L. Screening for BRCA1, BRCA2, CHEK2, PALB2, BRIP1, RAD50, and CDH1 mutations in high-risk Finnish BRCA1/2-founder mutation-negative breast and/or ovarian cancer individuals. Breast Cancer Res. 2011;13:R20.
Cybulski C, Lubiński J, Wokołorczyk D, Kuźniak W, Kashyap A, Sopik V, et al. Mutations predisposing to breast cancer in 12 candidate genes in breast cancer patients from Poland. Clin Genet. 2015;88:366–70.
Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–29.
Park J-Y, Zhang F, Andreassen PR. PALB2: the hub of a network of tumor suppressors involved in DNA damage responses. Biochim Biophys Acta. 1846;2014:263–75.
Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. 2009;19:524–9.
Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2007;39:159–61.
Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–4.
Foulkes WD, Ghadirian P, Akbari MR, Hamel N, Giroux S, Sabbaghian N, et al. Identification of a novel truncating PALB2 mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. 2007;9:R83.
Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–7.
Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia. Cancer Res. 2010;70:7353–9.
Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, et al. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. 2010;78:490–4.
García MJ, Fernández V, Osorio A, Barroso A, Llort G, Lázaro C, et al. Analysis of FANCB and FANCN/PALB2 fanconi anemia genes in BRCA1/2-negative Spanish breast cancer families. Breast Cancer Res Treat. 2009;113:545–51.
Dansonka-Mieszkowska A, Kluska A, Moes J, Dabrowska M, Nowakowska D, Niwinska A, et al. A novel germline PALB2 deletion in Polish breast and ovarian cancer patients. BMC Med Genet. 2010;11:20.
Bogdanova N, Sokolenko AP, Iyevleva AG, Abysheva SN, Blaut M, Bremer M, et al. PALB2 mutations in German and Russian patients with bilateral breast cancer. Breast Cancer Res Treat. 2011;126:545–50.
Janatova M, Kleibl Z, Stribrna J, Panczak A, Vesela K, Zimovjanova M, et al. The PALB2 gene is a strong candidate for clinical testing in BRCA1- and BRCA2-negative hereditary breast cancer. Cancer Epidemiol Biomarkers Prev. 2013;22:2323–32.
Blanco A, de la Hoya M, Osorio A, Diez O, Miramar MD, Infante M, et al. Analysis of PALB2 gene in BRCA1/BRCA2 negative Spanish hereditary breast/ovarian cancer families with pancreatic cancer cases. PLoS One. 2013;8, e67538.
Tischkowitz M, Sabbaghian N, Hamel N, Pouchet C, Foulkes WD, Mes-Masson A-M, et al. Contribution of the PALB2 c.2323C>T [p.Q775X] founder mutation in well-defined breast and/or ovarian cancer families and unselected ovarian cancer cases of French Canadian descent. BMC Med Genet. 2013;14:5.
Damiola F, Schultz I, Barjhoux L, Sornin V, Dondon M-G, Eon-Marchais S, et al. Mutation analysis of PALB2 gene in French breast cancer families. Breast Cancer Res Treat. 2015;154:463–71.
Thompson ER, Gorringe KL, Rowley SM, Wong-Brown MW, McInerny S, Li N, et al. Prevalence of PALB2 mutations in Australian familial breast cancer cases and controls. Breast Cancer Res. 2015;17:111.
Silvestri V, Rizzolo P, Zanna I, Falchetti M, Masala G, Bianchi S, et al. PALB2 mutations in male breast cancer: a population-based study in Central Italy. Breast Cancer Res Treat. 2010;122:299–301.
Southey MC, Teo ZL, Winship I. PALB2 and breast cancer: ready for clinical translation! Appl Clin Genet. 2013;6:43–52.
Noskowicz M, Bogdanova N, Bermisheva M, Takhirova Z, Antonenkova N, Khusnutdinova E, et al. Prevalence of PALB2 mutation c.509_510delGA in unselected breast cancer patients from Central and Eastern Europe. Fam Cancer. 2014;13:137–42.
Wojcik P, Jasiowka M, Strycharz E, Sobol M, Hodorowicz-Zaniewska D, Skotnicki P, et al. Recurrent mutations of BRCA1, BRCA2 and PALB2 in the population of breast and ovarian cancer patients in Southern Poland. Hered Cancer Clin Pract. 2016;14:5.
Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506.
Gaj P, Kluska A, Nowakowska D, Bałabas A, Piątkowska M, Dabrowska M, et al. High frequency of BRCA1 founder mutations in Polish women with nonfamilial breast cancer. Fam Cancer. 2012;11:623–8.
Kluska A, Balabas A, Paziewska A, Kulecka M, Nowakowska D, Mikula M, et al. New recurrent BRCA1/2 mutations in Polish patients with familial breast/ovarian cancer detected by next generation sequencing. BMC Med Genomics. 2015;8:19.
Lerner-Ellis J, Khalouei S, Sopik V, Narod SA. Genetic risk assessment and prevention: the role of genetic testing panels in breast cancer. Expert Rev Anticancer Ther. 2015;15:1315–26.
Tischkowitz M, Capanu M, Sabbaghian N, Li L, Liang X, Vallée MP, et al. Rare germline mutations in PALB2 and breast cancer risk: a population-based study. Hum Mutat. 2012;33:674–80.
Park J-Y, Singh TR, Nassar N, Zhang F, Freund M, Hanenberg H, et al. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene. 2014;33:4803–12.
Southey MC, Goldgar DE, Winqvist R, Pylkäs K, Couch F, Tischkowitz M, et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet. 2016. doi:10.1136/jmedgenet-2016-103839.
Catucci I, Radice P, Milne RL, Couch FJ, Southey MC, Peterlongo P. The PALB2 p.Leu939Trp mutation is not associated with breast cancer risk. Breast Cancer Res. 2016;18:111.
Acknowledgements
Not applicable.
Funding
This work was supported by the Ministry of Science and Higher Education (IP2012 041172, to MM).
Availability of data and material
The datasets generated and analyzed during the current study are not publicly available due to a concern to protect individual patient confidentiality (given the small number of PALB2 mutation carriers) but are available from the corresponding author on reasonable request.
Authors’ contributions
JO and MM proposed, designed the study and analyzed the data. AK, AB, MP, and KC prepared the DNA libraries, performed the sequencing and Taqman genotyping. KP analyzed sequencing data and performed statistical analyses. DN provided samples and analyzed clinical data. AK, MM, and JO drafted the manuscript. All authors have read and approve the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
The study included patients from the registry of the Genetic Counseling Unit, Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, who provided written informed consent for genetic testing at the initial counseling visit. Permission for genetic testing was obtained from the Local Ethical Committee of Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology on 9 May 2002 (No. 28/2002) and was subsequently extended to include genetic testing with next-generation sequencing on 11 June 2013 (No. 28/2002/1/2013) [29].
Consent for publication
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kluska, A., Balabas, A., Piatkowska, M. et al. PALB2 mutations in BRCA1/2-mutation negative breast and ovarian cancer patients from Poland. BMC Med Genomics 10, 14 (2017). https://doi.org/10.1186/s12920-017-0251-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-017-0251-8