
Abstract
Background
Lumpy skin disease (LSD) is a contagious viral disease of cattle caused by lumpy skin disease virus (LSDV). LSD has recently spread in Asia following outbreaks in the Middle East and Europe. The disease emerged in Bangladesh in July 2019 in the Chattogram district, then rapidly spread throughout the entire country. We investigated six LSD outbreaks in Bangladesh to record the clinical signs and collect samples for diagnostic confirmation. Furthermore, we performed the molecular characterization of Bangladesh isolates, analyzing the full RPO30 and GPCR genes and the partial EEV glycoprotein gene.
Results
Clinical observations revealed common LSD clinical signs in the affected cattle. PCR and real-time PCR, showed the presence of the LSDV genome in samples from all six districts. Phylogenetic analysis and detailed inspection of multiple sequence alignments revealed that Bangladesh isolates differ from common LSDV field isolates encountered in Africa, the Middle East, and Europe, as well as newly emerged LSDV variants in Russia and China. Instead, they were closely related to LSDV KSGP-0240, LSDV NI2490, and LSDV Kenya.
Conclusions
These results show the importance of continuous monitoring and characterization of circulating strains and the need to continually refine the strategies for differentiating vaccine strains from field viruses.
Similar content being viewed by others
Background
Lumpy skin disease (LSD) is a viral disease of cattle, caused by lumpy skin disease virus (LSDV) within the genus Capripoxvirus, family Poxviridae. The genus Capripoxvirus also comprises goatpox virus (GTPV) and sheeppox virus (SPPV). LSD is a notifiable disease by the World Organization for Animal Health (OIE) because of its potential rapid spread and substantial economic importance.
LSDV has a limited host range and does not infect non-ruminant hosts [1]. Both sexes and all ages of cattle breeds are susceptible to LSDV. However, younger animals may be more susceptible to the severe form of the disease [2]. Even in close contact with infected cattle, sheep and goats never developed LSD [3], with one noted exception, LSDVKSGP-0240 also known as LSDV KS1.
There is a significant variation of clinical signs with LSDV infections ranging from subclinical infection to death [4]. The main clinical signs include fever, the appearance of nodules in the skin, lesions in the mouth, pharynx, enlarged superficial lymph nodes, edema in the skin, and sometimes death [3,4,5]. There is an initial incubation period of 6 to 9 days during acute cases followed by a fever that may exceed 41 °C [6].
LSD is one of the most economically significant viral diseases of cattle because of the loss of production, permanent damage of hides, infertility, and death. Although the mortality rate is usually less than 10%, the disease morbidity rate can be as high as 100% [7].
For many years, the LSDV genome appeared to be stable. Indeed, following its first description in Zambia in 1929 [8], LSDV field isolates recovered for decades in Africa showed only minor genomic differences [9,10,11,12]. As the disease further spread into the Middle East from 2012 [13] and Europe in 2015 [7], the recovered LSDV field isolates showed little variability to contemporary African LSDV field isolates [14,15,16]. This genetic stability was exploited for the differentiation of LSDV live attenuated vaccines from contemporary field isolates [16,17,18,19,20,21].
However, this dynamic has shifted following the discovery of field LSDVs in Russia in 2017 and 2019 showing vaccine-like profiles [22,23,24,25]. Some of these LSDV variants, presented a 12-nucleotide insertion in the GPCR gene, like vaccine strains, and others presented a27-nucleotide deletion, similar to the LSDV Neethling vaccine strain, in the ORF LSDV 126. The authors attributed these variants’ emergence to recombination events between the Neethling vaccine strain and field isolates [23]. This has prompted researchers [26] to question the relevance of current strategies to differentiate LSDV vaccine strains from viral field strains. Similarly, the LSDV strains involved in the outbreaks in China present the GPCR profile of LSDV vaccines with the 27-nucleotide insertion in their EEV glycoprotein gene. Interestingly, Kononova et al. [27], also showed in vitro and in vivo that the recombinant LSDVs could induce more severe disease than the typical field isolates.
The increased variability of LSDV in recent years makes it crucial to adapt the molecular DIVA strategies based on the knowledge of the circulating strain of LSDV. This requires the constant monitoring and characterization of LSDV field isolates.
Several PCR, real-time PCR, and HRM based methods are available for the detection of the LSDV genome, [12, 14, 28,29,30,31,32,33,34,35,36] and molecular epidemiological studies of LSDV rely on analyzing various genomic regions, such as the GPCR, the RPO30, the P32, and the EEV glycoprotein genes [11, 12, 17,18,19, 37].
On September 15, 2019, Bangladesh notified to OIE the first outbreak of LSD in the country. The disease started in July 2019 in the Southeast (Chattogram district) of the country, then progressively spread throughout the country. Because of the wide distribution and large cattle population in Bangladesh, LSD is now one of the most economically important emerging livestock disease in Bangladesh.
This study aimed to investigate and confirm the recent outbreaks and provide LSDV molecular characterization in different regions in Bangladesh.
Results
Outbreak investigation
All affected cattle in different districts in Bangladesh (Chottogram, Dhaka, Gazipur, Narayanganj, Pabna, and Satkhira) showed the following common clinical signs: fever (40–41 °C), depression, loss of appetite, nasal and ocular discharges, salivation, circumscribed nodules with different sizes on the skin covering their head, neck, trunk, perineum, udder, and teats. Figure 1 illustrates the skin lesions of affected cattle. In many infected animals, the necrotic nodules were ulcerated and formed deep scabs (sitfast). Moreover, a decrease in body weight and reduced milk production were prominent signs observed in cattle affected by LSD. The total cattle population, reported morbidities and mortalities in the six districts of this study (Fig. 2) are sumarised in Table 1.

Skin lesions characteristics of lumpy skin disease in 3 animals in Bangladesh. The generalized circumscribed active nodular skin lesions covering the entire body are visible. Source: own

Map of Bangladesh showing the sample collection area. The map is an own creation using Arc GIS software version 13.2
Molecular detection of LSDV
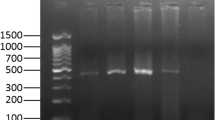
Gel electrophoresis of the P32 amplicons showed a 390 bp product in all fifty (50) samples collected in six districts, as illustrated for selected samples in Fig. 3.

Agarose gel electrophoresis showing the 390 bp amplicon of P32 gene for selected samples of Bangladesh. Lane M: 100 bp DNA ladder, Lane 1–5: LSDV field samples, Lane PC: positive control, lane NC: negative control
The real-time PCR result confirmed capripoxvirus DNA in all samples. Six representative samples, one per district with a Cq value between 19.17 and 25.31, were selected for sequencing (Table 2).
Amplification and sequencing of the RPO30, GPCR, and EEV glycoprotein genes
We have successfully amplified and sequenced two fragments for the RPO30 gene (554 bp and 520 bp) in 6 samples and three for the GPCR (617 bp, 603 bp, and 684 bp) in 5 samples out of 6. We also amplified and sequenced the partial EEV glycoprotein gene in 6 samples. The complete RPO30 and GPCR genes and the partial EEV glycoprotein gene sequences were submitted to the GenBank database under accession numbers MT448690 to MT448701.
Phylogenetic analysis
For each of the targeted genes, the sequences of the Bangladesh LSDVs showed 100% identity among each other. On the phylogenetic trees for both RPO30 (Fig. 4) and GPCR (Fig. 5), all the Bangladesh LSDVs clustered together.

Maximum clade credibility (MCC) tree based on the complete RPO30 complete gene sequences of capripoxviruses. The posterior probabilities are plotted as respective nodes labels. LSDVs from Bangladesh are highlighted in red and reference sequences are represented with their accession numbers

Maximum clade credibility (MCC) tree based on the complete GPCR gene sequences of Capripoxviruses, plotted together with multiple sequence alignment. Only the portion of the alignment between positions 80 and 120 is shown. The posterior probabilities are plotted as respective nodes labels. LSDVs from Bangladesh are highlighted in red and reference sequences are represented with their accession numbers
On the RPO30 tree (Fig. 4), Bangladesh isolates clustered within subgroup I, tightly with LSDV KSGP 0240 (KX683219), known as LSDV KS1, LSDV NI-2490 (AF325528), Indian LSDV field isolates, and two recombinant LSDV field isolates from Russia, LSDV Russia/Udmurtiya/2019 (MT134042), and LSDV Russia/Saratov/2017 (MH646674). The commonly circulating field isolates from Africa, the Middle East, and Europe are segregated from the Bangladesh isolates, clustering within subgroup II. A third subgroup contained mainly LSDV Neethling derived vaccine strains, the historical field LSDV RSA/54 Haden, and the LSDV field isolates from China. On the GPCR tree (Fig. 5), there were only two sub-groups. Bangladesh LSDV isolates clustered within subgroup I, together with LSDV SGP_O-240 (KJ818288), LSDV NI-2490 (AF325528), LSDV Kenya (MN072619), common LSDV field isolates from Africa, the Middle East, and Europe, LSDV Xinjiang/2019 (MN598006), and LSDV China/XJ/2019(MN508357) from China. The second subgroup of the GPCR consisted of LSDV Neethling derived vaccines, LSDV RSA/54 Haden (FJ869376), and three recombinant LSDVs from Russia: LSDV Russia/Udmurtiya/2019 (MT134042), LSDV Russia/Saratov/2017 (MH646674) and LSDV Dergachevskyi (MH029290). The multiple sequence alignments of the GPCR gene showed that the Bangladesh LSDV contained the 12-nucleotide insertion (Fig. 5). This 12-nucleotide insertion is also present in the two common LSDV vaccine strains (LSDV KSGP 0240 and LSDV Neethling) and a few historical field isolates (collected before 1960) such as LSDV NI-2490 (AF325528), LSDV Kenya (MN072619), and LSDV RSA/54Haden (GU119937). This insertion is also present in recombinant LSDVs from Russia (LSDV Russia/Udmurtiya/2019, LSDV Russia/Saratov/2017, and LSDV Dergachevskyi) and in recent LSDV isolates from China.
Alignment of the EEV glycoprotein gene sequence showed a 27-nucleotide insertion in all LSDVs from Bangladesh (Fig. 6), which is characteristic of common field isolates and also present in the LSDV KSGP-0240 derived vaccines and historical LSDVs, LSDV NI2490 (1958) and LSDV Kenya (1950), both from Kenya.

Multiple sequence alignments of the partial nucleotide sequences of the of EEV glycoprotein gene. LSDVs from Bangladesh were aligned with representative LSDVs’ sequences retrieved from GenBank. A unique sequence signature of 27-nucleotide only in LSDV Neethling like viruses is highlighted in the box. Identical nucleotides are indicated with dots
Taken together, the analyses of all three targets suggest that the Bangladesh LSDVs were more related to LSDV KSGP-0240, LSDV NI-2490, and LSDV Kenya. They differed from all recent LSDV field isolates, including the LSDVs from China and the recombinant LSDVs described in Russia, and LSDV Neethling vaccine strain.
Discussion
The diagnosis of LSD was confirmed by real time PCR and the viruses in samples collected from outbreaks between July and September 2019 in Bangladesh were molecularly characterized.
LSD emerged in Bangladesh in July 2019, hitting the district of Chattogram, before quickly spreading to Dhaka, Naryanganj, Gazipur, Satkhira, and Pabna regions, between July and September 2019 [38]. The affected cattle exhibited common clinical manifestations of LSD in cattle, including nasal and ocular discharges and skin lesions [39]. The epidemiology unit data shows a very high incidence of LSD in the Chattogram province compared to other regions in the country. A plausible explanation is that Chattogram, a port city of Bangladesh, is a major coastal city and financial center in southeastern Bangladesh, with more cattle movement due to trade. It was also the first affected province.
The Bangladesh LSDVs presented the 12-nucleotide insertion found in the GPCR gene of LSDV KSGP-0240, LSDV Neethling vaccines, and a few historical LSDVs such as the LSDV NI2490 and LSDV Kenya, LSDVs from China and the recombinant LSDVs described in Russia. The presence of this 12-nucleotide insertion makes them different from commonly circulating field LSDVs encountered in Africa, Europe, and the Middle East [11, 16, 19, 24, 40]. However, a 27-nucleotide insertion in the EEV glycoprotein gene of Bangladesh isolates, and the RPO30 and GPCR gene trees’ analysis differentiated them from LSDV Neethling derived vaccines. This 27-nucleotide insertion in the EEV glycoprotein also makes them different from Chinese LSDV isolates as well as from the recombinant LSDVs described in Russia in recent years.
A close inspection of the sequence alignment of the EEV glycoprotein, the GPCR, and RPO30 genes showed 100% identity to the LSDV KSGP 0240, LSDV NI2490, and LSDV Kenya at the nucleotide level. These features make them unique, as the commonly circulating LSDV isolates have not demonstrated that level of closeness to LSDV KSGP 0240, LSDV NI2490, and LSDV Kenya. It is worth noting that the Bangladesh LSDV RPO30 sequence was 100% identical with the Indian isolate, however, as no GPCR and EEV glycoprotein gene sequences were available from India, it was not possible to extend the comparison.
The existence of vaccine-like field isolates with mixed characteristics between common field viruses and the LSDV Neethling vaccine has been reported in Russia [24]. A more recent report described a field LSDV isolate in Kurgan, Russia, exhibiting similarities to LSDV KSGP 0240 and LSDV NI2490 based on the analysis of GPCR and RPO30 gene fragments [26]. Although the complete RPO30 and GPCR sequences were not available for a full comparison, we noticed a nucleotide difference between the partial RPO30 sequence of the Kurgan isolate and those of this study. Our findings support the circulation of LSDV KS1 or LSDV NI2490-like virus in the field.
How such a virus has emerged suddenly in Bangladesh remains unknown. An extensive characterization of LSDV in neighboring countries could help resolve the emergence of these isolates.
Previous studies showed that the use of the LSDV KSGP 0240 for vaccination could lead to the appearance of generalized lesions [15, 41]. The lesions in cattle in Bangladesh showed pathogenic virus-like lesions, especially the presence of deep “sit fast,” not usually observed with KSGP 0240-induced disease [15]. It is also worth noting that Bangladesh was not vaccinating cattle against LSD before these outbreaks but later started vaccination using a goat poxvirus strain. Therefore, it is unlikely that the administration of a good quality LSDV KSGP 0240 vaccine caused these outbreaks. Furthermore, LSDV KSGP 0240-induced disease manifests only as an adverse reaction in vaccinated animals and shows no signs of animal to animal spread [15, 41].
Historically, viruses resembling LSDV KSGP 0240, the LSDV NI2490 (1958), and LSDV Kenya (1950, but sequenced only recently), caused LSD outbreaks in Kenya [10]; however, these viruses were never detected in subsequent LSDV epidemics in Africa, the Middle East, and Europe. Whether Bangladesh isolates, and presumably those described in Kurgan, Russia, relate to LSDV NI2490 and LSDV Kenya is unclear. Further investigation through full genome sequencing is warranted, as none of the three targeted genes of this study could provide differentiation between LSDV KSGP-0240 and LSDV NI2490.
The reason for the emergence of such LSDV variants remains uncertain. However, recent reports from Russia suggest the possibility of recombination events [24].
Conclusions
In conclusion, using a multi-targeted approach,we discovered that the viruses causing outbreaks in Bangladesh were different from common contemporary LSDV field isolates circulating worldwide, including the Chinese isolates and the recombinant LSDVs described between 2017 and 2019 in Russia. Full genome analysis will elucidate whether these viruses are LSDV KSGP-0240 or LSDV NI2490/LSDV Kenya. This study highlights the importance of continuous monitoring and characterization of circulating strains and the need to continually refine the strategies for differentiating vaccine strains from field viruses.
Methods
Outbreak investigations and sample collection
Fifty (50) biopsies of skin nodules were collected from 6 different districts of Bangladesh (Fig. 2) between July and September 2019. Table 2 shows the location, source, and collection period of LSDV samples. The samples were collected aseptically and transported in a cool box to the Central Disease Investigation Laboratory (CDIL) at Dhaka, Bangladesh. Samples were stored at − 80 °C for further processing.
Sample preparation and DNA extraction
Biopsy nodule samples were cut with a scalpel into small pieces. Pieces were macerated with pestle and mortar, then transferred to sterile tubes with 10 ml sterile phosphate-buffered saline (PBS) to prepare tissue homogenates. Tubes were centrifuged at 1000 g, and 200 μl of supernatant was transferred to an Eppendorf tube for DNA extraction.
DNA extraction from skin samples was performed using DNeasy Blood & Tissuekit (Qiagen, Germany) according to the manufacturer’s recommendations. The DNA was eluted using 70 ul elution buffer and stored at − 20 °C until further use.
Molecular detection
A conventional PCR was carried out to amplify a 390 bp fragment within the P32 gene of capripoxviruses [28]. The PCR was performed using the Platinum™ Taq DNA Polymerase kit (cat# 10966–026) in a reaction volume of 25 μl containing 2.5 μl 10 X PCR buffer, 0.75 μl Magnesium chloride (50 mM), 0.5 ul dNTPs (10 mM), 0.1 μl Platinum Taq DNA polymerase, 400 nM of each primer and 1 μl template DNA. The PCR tubes containing the above mixture were transferred into a thermal cycler (T-1000, Bio-Rad, USA), and amplification was conducted with the following program: initial denaturation at 94 °C for 5 min, 38 cycles denaturation at 94 °C for 30 s, annealing at 50 °C for 30 s, extension at 72 °C for 30 s; and a final extension phase run at 72 °C for 5 min.
The PCR products were separated by gel electrophoresis on a 1.5% agarose for 60 min and visualized with a gel documentation system (UVP GelStudio PLUS Gel Documentation Imaging Systems, Analytik Jena, Germany).
A real-time PCR for the detection of capripoxvirus DNA was performed as previously described [31] with some modifications.
Briefly, the PCR mixture was set up in a reaction volume of 25 μl, containing 12.5 μl of the iQsupermix (Bio-Rad, USA), 400 nM of each primer, 250 nM of the fluorogenic probe and 5 μl of template. The reaction consisted of an initial denaturation step at 95 °C for 10 min, followed by 45 cycles at 95 °C for 15 s and 60 °C for 60 s with the fluorescence recording at the end of the combined annealing elongation step. The assaywasperformed using the CFX real-time PCR detection system (Bio-Rad).
Amplification and sequencing of the RPO30, GPCR, and EEV glycoprotein genes
The RPO30 and the GPCR were amplified as previously described [19].
A pair of primers; EEVGly F- 5′- ATGGGAATAGTATCTGTTGTATACG-3′ and EEVGly R-5′- CGAACCCCTATTTACTTGAGAA-3′ were designed for the amplification of fragments containing the partial EEV glycoprotein (encoded by ORF LSDV126) and hypothetical protein LSDV 127 gene [18]. The PCR reaction was performed in a reaction volume of 20 μl containing 500 nM of each of the forward and reverse primers, 0.2 mM of dNTPs, 1x buffer (Qiagen), 2.5 U of Taq DNA polymerase (Qiagen), and 2 μl template DNA. The amplification consisted of an initial denaturation at 95 °C for 4 min followed by 35 cycles of 95 °C for 40 s, 55 °C for 30 s, and 72 °C for 1 min, and a final extension step at 72 °C for 7 min.
The PCR products were separated by electrophoresis on a 1.5% agarose gel at 100 V for 60 min and visualized using a Gel Documentation System (Bio-Rad, USA).
The PCR amplicons were purified using the Wizard SV Gel and PCR clean-up system kit (Promega) according to the manufacturer’s instructions. LGC Genomics (Germany) performed the sequencing of the purified PCR amplicons. Vector NTI 11.5 software (Invitrogen, USA) was used for sequencing data analysis and assembly.
Phylogenetic analysis
Nucleotide sequences were aligned using the Muscle algorithm and the codon option implemented in MEGA7 [42]. The complete RPO30 and GPCR gene sequences of additional CaPVs (LSDVs, GTPVs, and SPPVs), retrieved from GenBank, were included for comparative analyses.
The file with aligned sequences in FASTA was converted to a Nexus format using Seaview. The Bayesian phylogenetic inference was performed with BEAST v1.8.4 [43]. First, the BEAUti module was used to generate BEAST files using the HKY substitution +G nucleotide substitution and a UPGMA starting tree option. The Markov Chain Monte Carlo method was run with BEAST, for 10,000,000 generations with a sample taken each 10,000 generations. The TRACER program was used to inspect the log files and determine the optimum number of burn-in based on the Effective Sample Sizes (ESS > 200).
TreeAnnotator was used to generate the Maximum Clade Credibility (MCC) after discarding the 3% burn-in. The tree was visualized with the associated meta-data using the ggtree package in R [44]. Additionally, for the GPCR tree, the multiple sequence alignment file of the nucleotide sequences was imported. A specific slice of the alignment, between positions 80 and 120, was visualized together with the tree [44].
Availability of data and materials
DNA sequences generated and analyzed under the current study are available in GenBank under accession numbers MT448690 to MT448701. All the remaining datasets generated during this study are available from the corresponding author on request.
Abbreviations
- CDLI:
-
Central Disease Investigation Laboratory
- DNA:
-
Deoxyribonucleic acid
- dNTP:
-
Deoxynucleotide triphosphate
- EEV:
-
Extracellular enveloped virus
- ESS:
-
Effective Sample Sizes
- GPCR:
-
G protein-coupled receptor
- GTPV:
-
Goatpox virus
- HRM:
-
High-resolution melting
- LSD:
-
Lumpy skin disease
- LSDV:
-
Lumpy skin disease virus
- MCC:
-
Maximum clade credibility
- OIE:
-
World Organization for Animal Health
- ORF:
-
Open reading frame
- PBS:
-
Phosphate-buffered saline
- PCR:
-
Polymerase chain reaction
- RPO30:
-
RNA polymerase 30 kDa subunit
- SPPV:
-
Sheeppox virus
References
Shen YJ, Shephard E, Douglass N, Johnston N, Adams C, Williamson C, et al. A novel candidate HIV vaccine vector based on the replication deficient Capripoxvirus, lumpy skin disease virus (LSDV). Virol J. 2011;8:1–2.
Tageldin MH, Wallace DB, Gerdes GH, Putterill JF, Greyling RR, Phosiwa MN, et al. Lumpy skin disease of cattle: an emerging problem in the Sultanate of Oman. Trop Anim Health Prod. 2014;46:241–6.
Davies FG. Lumpy skin disease, an African capripox virus disease of cattle. Br Vet J. 1991;147:489–503.
Carn VM, Kitching RP. An investigation of possible routes of transmission of lumpy skin disease virus (Neethling). Epidemiol Infect. 1995;114:219–26.
OIE. Lumpy skin disease. In: Manual of diagnostic tests and vaccines for terrestrial animals; 2017. http://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/2.04.13_LSD.pdf.
USDA. Lumpy skin disease standard operating procedures: 1. overview of etiology and ecology. 2016. https://www.aphis.usda.gov/animal_health/emergency_management/downloads/sop/lsdv_fadprep_ee.pdf. Accessed 19 July 2020.
Tuppurainen ESM, Venter EH, Shisler JL, Gari G, Mekonnen GA, Juleff N, et al. Review: Capripoxvirus diseases: current status and opportunities for control. Transbound Emerg Dis. 2017;64:729–45.
Tuppurainen ESM, Stoltsz WH, Troskie M, Wallace DB, Oura CAL, Mellor PS, et al. A potential role for Ixodid (hard) tick vectors in the transmission of lumpy skin disease virus in cattle. Transbound Emerg Dis. 2011;58:93–104.
Kara PD, Afonso CL, Wallace DB, Kutish GF, Abolnik C, Lu Z, et al. Comparative sequence analysis of the south African vaccine strain and two virulent field isolates of lumpy skin disease virus. Arch Virol. 2003;148:1335–56.
Tulman ER, Afonso CL, Lu Z, Zsak L, Kutish GF, Rock DL. Genome of lumpy skin disease virus. J Virol. 2001;75:7122–30.
Le Goff C, Lamien CE, Fakhfakh E, Chadeyras A, Aba-Adulugba E, Libeau G, et al. Capripoxvirus G-protein-coupled chemokine receptor: a host-range gene suitable for virus animal origin discrimination. J Gen Virol. 2009;90:1967–77.
Lamien CE, Le Goff C, Silber R, Wallace DB, Gulyaz V, Tuppurainen E, et al. Use of the Capripoxvirus homologue of Vaccinia virus 30 kDa RNA polymerase subunit (RPO30) gene as a novel diagnostic and genotyping target: development of a classical PCR method to differentiate goat poxvirus from sheep poxvirus. Vet Microbiol. 2011;149:30–9.
Alkhamis MA, VanderWaal K. Spatial and temporal epidemiology of lumpy skin disease in the Middle East, 2012–2015. Front Vet Sci. 2016;3:19.
Stram Y, Kuznetzova L, Friedgut O, Gelman B, Yadin H, Rubinstein-Guini M. The use of lumpy skin disease virus genome termini for detection and phylogenetic analysis. J Virol Methods. 2008;151:225–9.
Abutarbush SM, Hananeh WM, Ramadan W, Al Sheyab OM, Alnajjar AR, Al Zoubi IG, et al. Adverse reactions to field vaccination against lumpy skin disease in Jordan. Transbound Emerg Dis. 2016;63:e213–9.
Agianniotaki EI, Tasioudi KE, Chaintoutis SC, Iliadou P, Mangana-Vougiouka O, Kirtzalidou A, et al. Lumpy skin disease outbreaks in Greece during 2015–16, implementation of emergency immunization and genetic differentiation between field isolates and vaccine virus strains. Vet Microbiol. 2017;201:78–84.
Menasherow S, Rubinstein-Giuni M, Kovtunenko A, Eyngor Y, Fridgut O, Rotenberg D, et al. Development of an assay to differentiate between virulent and vaccine strains of lumpy skin disease virus (LSDV). J Virol Methods. 2014;199:95–101.
Menasherow S, Erster O, Rubinstein-Giuni M, Kovtunenko A, Eyngor E, Gelman B, et al. A high-resolution melting (HRM) assay for the differentiation between Israeli field and Neethling vaccine lumpy skin disease viruses. J Virol Methods. 2016;232:12–5.
Gelaye E, Belay A, Ayelet G, Jenberie S, Yami M, Loitsch A, et al. Capripox disease in Ethiopia : Genetic differences between field isolates and vaccine strain , and implications for vaccination failure. Antivir Res. 2015;119:28–35.
Agianniotaki EI, Chaintoutis SC, Haegeman A, Tasioudi KE, De Leeuw I, Katsoulos PD, et al. Development and validation of a TaqMan probe-based real-time PCR method for the differentiation of wild type lumpy skin disease virus from vaccine virus strains. J Virol Methods. 2017;249:48–57.
Pestova Y, Byadovskaya O, Kononov A, Sprygin A. A real time high-resolution melting PCR assay for detection and differentiation among sheep pox virus, goat pox virus, field and vaccine strains of lumpy skin disease virus. Mol Cell Probes. 2018;41:57–60.
Kononov A, Byadovskaya O, Kononova S, Yashin R, Zinyakov N, Mischenko V, et al. Detection of vaccine-like strains of lumpy skin disease virus in outbreaks in Russia in 2017. Arch Virol. 2019;164:1575–85.
Sprygin A, Pestova Y, Bjadovskaya O, Prutnikov P, Zinyakov N, Kononova S, et al. Evidence of recombination of vaccine strains of lumpy skin disease virus with field strains, causing disease. PLoS One. 2020;15:e0232584.
Sprygin A, Babin Y, Pestova Y, Kononova S, Wallace DB, Van Schalkwyk A, et al. Analysis and insights into recombination signals in lumpy skin disease virus recovered in the field. PLoS One. 2018;13:e0207480.
Sprygin A, Pestova Y, Prutnikov P, Kononov A. Detection of vaccine-like lumpy skin disease virus in cattle and Musca domestica L. flies in an outbreak of lumpy skin disease in Russia in 2017. Transbound Emerg Dis. 2018;65:1137–44.
Aleksandr K, Pavel P, Olga B, Svetlana K, Vladimir R, Yana P, et al. Emergence of a new lumpy skin disease virus variant in Kurgan oblast, Russia, in 2018. Arch Virol. 2020;165:1343–56.
Kononova S, Kononov A, Shumilova I, Byadovskaya O, Nesterov A, Prutnikov P, et al. A lumpy skin disease virus which underwent a recombination event demonstrates more aggressive growth in primary cells and cattle than the classical field isolate. Transbound Emerg Dis. 2020;00:1–7. https://doi.org/10.1111/tbed.13798.
Heine HG, Stevens MP, Foord AJ, Boyle DB. A capripoxvirus detection PCR and antibody ELISA based on the major antigen P32, the homolog of the vaccinia virus H3L gene. J Immunol Methods. 1999;227:187–96.
Ireland DC, Binepal YS. Improved detection of capripoxvirus in biopsy samples by PCR. J Virol Methods. 1998;74:1–7.
Balinsky CA, Delhon G, Smoliga G, Prarat M, French RA, Geary SJ, et al. Rapid preclinical detection of sheeppox virus by a real-time PCR assay. J Clin Microbiol. 2008;46:438–42.
Bowden TR, Babiuk SL, Parkyn GR, Copps JS, Boyle DB. Capripoxvirus tissue tropism and shedding: a quantitative study in experimentally infected sheep and goats. Virology. 2008;371:380–93.
Stubbs S, Oura CAL, Henstock M, Bowden TR, King DP, Tuppurainen ESM. Validation of a high-throughput real-time polymerase chain reaction assay for the detection of capripoxviral DNA. J Virol Methods. 2012;179:419–22.
Haegeman A, Zro K, Vandenbussche F, Demeestere L, Van Campe W, Ennaji MM, et al. Development and validation of three Capripoxvirus real-time PCRs for parallel testing. J Virol Methods. 2013;193:446–51.
Lamien CE, Lelenta M, Goger W, Silber R, Tuppurainen E, Matijevic M, et al. Real time PCR method for simultaneous detection, quantitation and differentiation of capripoxviruses. J Virol Methods. 2011;171:134–40.
Gelaye E, Lamien CE, Silber R, Tuppurainen ESM, Grabherr R, Diallo A. Development of a cost-effective method for Capripoxvirus genotyping using snapback primer and dsDNA intercalating dye. PLoS One. 2013;8:e75971.
Gelaye E, Mach L, Kolodziejek J, Grabherr R, Loitsch A, Achenbach JE, et al. A novel HRM assay for the simultaneous detection and differentiation of eight poxviruses of medical and veterinary importance. Sci Rep. 2017;7:42892.
Hosamani M, Mondal B, Tembhurne PA, Bandyopadhyay SK, Singh RK, Rasool TJ. Differentiation of sheep pox and goat poxviruses by sequence analysis and PCR-RFLP of P32 gene. Virus Genes. 2004;29:73–80.
Anonymous. Situation Report: Lumpy Skin Disease in Bangladesh Background. 2019. https://www.oie.int/fileadmin/Home/eng/Animal_Health_in_the_World/docs/pdf/Disease_cards/LUMPY_SKIN_DISEASE_FINAL.pdf. Accessed 19 July 2020.
Babiuk S, Bowden TR, Parkyn G, Dalman B, Manning L, Neufeld J, et al. Quantification of lumpy skin disease virus following experimental infection in cattle. Transbound Emerg Dis. 2008;55:299–307.
Şevik M, Avci O, Doǧan M, Ince ÖB. Serum biochemistry of lumpy skin disease virus-infected cattle. Biomed Res Int. 2016;2016:6257984.
Yeruham I, Perl S, Nyska A, Abraham A, Davidson M, Haymovitch M, et al. Adverse reactions in cattle to a capripox vaccine. Vet Rec. 1994;135:330–2.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Yu G, Smith DK, Zhu H, Guan Y, Lam TTY. Ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8:28–36.
Acknowledgments
We are very grateful to the veterinarian of the Department of Livestock Services, who helped a lot in the sample collection.
Funding
This study was supported by VETLAB network initiative of the Joint FAO/IAEA Division, funded through the African Renaissance and International Cooperation fund of South Africa and the Peaceful Uses Initiatives (PUI) by Japan and the United States of America. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: SCB, MS, IAK; Performed the experiments: SCB, MGAC, TBKS, SA; Analyzed the data: CEL, TBKS, SA; Contributed reagents/materials/analysis tools: MAUF, MGAC, TABMMGO, FT; Wrote the paper: MS, CEL, NB; Supervised the study. MBR, GC, IAK; Edited the final manuscript: TABMMGO, FT, NB, MBR, GC, MAUF. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval of this study was obtained through the approval of Central Disease Investigation Laboratory (CDIL), Bangladesh. The samples were summited to the Central Disease Investigation Laboratory (CDIL) for diagnostic confirmation and the results reported to the OIE. Sampling was carried out under the owner’s consent.
Consent for publication
Not applicable.
Competing interests
All authors declared that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Badhy, S.C., Chowdhury, M.G.A., Settypalli, T.B.K. et al. Molecular characterization of lumpy skin disease virus (LSDV) emerged in Bangladesh reveals unique genetic features compared to contemporary field strains. BMC Vet Res 17, 61 (2021). https://doi.org/10.1186/s12917-021-02751-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-021-02751-x