
Abstract
Background
Porcine circovirus 2-associated disease (PCVAD) is acknowledged as one of the most economically important diseases for the swine industry worldwide. The aim of this study was to characterize and determine the genetic diversity of PCV2 in the porcine population of Guangxi, China.
Methods
The full length genome and open reading frame 2 (ORF2) of 95 PCV2 strains collected from the tissues and sera of pigs that had either died as a result of PCVAD or did not exhibit disease symptoms were analyzed.
Results
The results of multiple sequence alignments showed that there is considerable diversity among the PCV2 ORF2 sequences. Phylogenetic analyses based on the complete genome showed that current PCV2 strains in this study could be divided into PCV2a (1/95), PCV2b (39/95), PCV2d (43/95), PCV2e (10/95) and PCV2h (2/95). Among the 5 sub-genotypes, PCV2b was dominant in the porcine population from 2002 to 2008. The newly identified sub-genotype, PCV2d, was seen from 2003 and has increased every year. PCV2b and PCV2d formed two predominant genetic groups circulating in southern China between 2009 and 2013 and the sub-genotype PCV2d has become the dominant virus in China since 2014.
Conclusions
This study reveals the complex genetic diversity of PCV2 and improves our understanding regarding the epidemiological trends of PCV2 sub-genotypes in China.
Similar content being viewed by others


Background
Porcine circovirus 2 (PCV2) is the major etiological agent that causes PCV2-associated diseases (PCVAD) in growing pigs. This includes post-weaning multi-systemic wasting syndrome (PMWS), porcine dermatitis and nephropathy syndrome (PDNS), porcine respiratory disease complex (PRDC), congenital tremors type II (CT) and reproductive failure [1,2,3,4]. PCV2 is a small, single-stranded, non-enveloped, circular DNA virus containing a genome of 1766–1768 nt [5]. The PCV2 genome contains 11 open reading frames (ORFs) [5]. Five proteins, encoded by ORF1 to ORF5, are currently studied and recognized as the functional proteins of PCV2 [6,7,8,9,10,11]. Among these, the main ORF1 and ORF2 were identified as genes encoding viral replicase (Rep and Rep’) and capsid protein, respectively [6, 11].
It has been shown that PCV2 is continuously evolving through point mutation and genome recombination, which can lead to some new antigenic variant strains and it is known that new PCV2 variant strains are emerging [12,13,14]. Phylogenetic analyses of the complete genome and ORF2 region of PCV2 isolates worldwide have shown that PCV2 could be divided into eight distinct genotypes. These have been named PCV2a, PCV2b, PCV2c, PCV2d, PCV2e, PCV2f, PCV2g and PCV2h according to a new genotyping methodology protocol [15]. PCV2a, PCV2b and PCV2d have been circulating worldwide and shown to have five (2A–E), three (1A–C) and two sub-genotypes (2d-1 and 2d-2) [13, 16], respectively, while the presence of PCV2c has only been reported in Denmark and Brazil [16,17,18]. PCV2e has been identified in pigs from China, Thailand, USA and Mexico [19,20,21,22]. Amongst all PCV2 genotypes, PCV2a was the predominant strain prior to 2000 and then there appeared to be a global genetic shift from PCV2a to PCV2b with the latter being the predominant genotype seen in the past ten years [18, 19, 23, 24]. Recently, there is a number of reports which suggest that there is an ongoing genotype shift occurring from PCV2b to PCV2d [13, 25]. In 2010, a variant PCV2 mutant strain designated mPCV2b, now grouped in PCV2d, with an elongation of its ORF2 by one amino acid, lysine (K), was identified in several PCVAD cases in China and other countries and recent studies showed that the prevalence rate of mPCV2b appears to have increased in China and a similar trend is evident in the U.S.A [13, 23, 26, 27].
Although there is increasing use of killed or subunit vaccines against PCV2 in pigs, the prevalence of PCV2 in China is still on the rise. Guangxi Province is one of the biggest pig breeding regions in China. The aim of this study was to investigate the prevalence and genetic variation of PCV2 in China using strains observed in the pig population from 2002 to 2016. Our findings revealed that PCV2d has becoming the predominating virus since 2014. Overall, this study helps to elucidate important aspects of the molecular genetic evolution of PCV2 and this is a prerequisite for the future development of effective disease control and prevention strategies for the spread of this virus.
Results
Prevalence of PRRSV in Guangxi Province, China from 2002 to 2016
Of the 371 filed samples collected from the clinical diseased and health pigs between 2002 and 2016 in the Guangxi Province of China, 181 samples (48.8%) were positive for PCV2, as determined by specific PCR. These results indicate that PCV2 is distributed widely among swine populations in the Guangxi Province.
Sequence and phylogenetic analyses of the ORF2 gene of PCV2
To explore the genetic relationship and evolution of PCV2 from 2002 to 2016, 95 of 181 PCV2 positive samples were used for genome amplification and sequencing and phylogenetic analysis was carried out based on the sequences of the ORF2 gene of 95 PCV2 isolates with reference sequences. The results showed that the complete genomes of all 95 strains were 1767 or 1768 bp in length as shown in Table 1. Forty of the 95 ORF2 nucleotide sequences were 702 bp in length, encoding a Cap protein of 233 amino acid residues. Forty five of the 95 ORF2 nucleotide sequences were 705 bp, encoding a Cap protein of 234 amino acid residues. These strains are also known as mutant PCV2 (mPCV2), which has a codon shift from TTA to CTT in ORF2, resulting in a mutation of the stop codon (from UAA to AAG) in the ORF2, leading to an extended lysine (K) residue encoded by AAG or AAA.
Comparisons of the complete genomic sequence revealed 96.6% identity between PCV2a strains and the reference PCV2a strains (Table 2). The nucleotide sequence identity between PCV2b strains and reference PCV2b strains was 97.0–99.4%. The nucleotide sequence identity between PCV2d strains and the reference PCV2d strains was 97.1–99.9% and the nucleotide sequence identity between PCV2e strains and the reference PCV2e strains was 97.6~99.4% (Table 2). To investigate variations in the deduced amino acid sequences of ORF2 gene products, the amino acid sequences of 95 PCV2 strains including some representative strains were aligned. The results showed that there are five major regions of variation among the PCV2 strains. These include residues 57–91, 121–151, 181–191, 206–215 and 230–233 (Fig.1). One of the 96 strains has a typical TNKISI motif present in PCV2a. 39 of the 96 strains have typical S/PNPRSV and A/TGIE motifs present in PCV2b and 43/95 strains have SNPLTV and TGID motifs present in most of the PCV2d. PCV2e strains have a typical TNKISI motif which are also present in the PCV2a strains. Compared with PCV2a, PCV2e have specific substitutions at positions 47 (T to S), 72 (R to L), 131 (P to F), 187 (L to I) and 191(R to K). PCV2h strains have a typical SNPLTV motif which is present in most of the PCV2d strains. But the motif, TGID, was changed to SAID. Specific aa changes in the reported antibody epitope regions and immune-dominant decoy epitope regions (57–91, 181–191 and 230–233) of the Cap protein were found in some strains. Moreover, specific aa changes at positions 133–135 were also identified in some strains. As a result of a mutation at the stop codon, 45 of the 96 strains had an extended lysine (K) residue encoded by AAG or AAA.

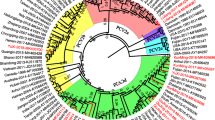
Phylogenetic tree based on a comparison of 129 complete PCV2 genomic sequences, including the 95 sequences from this study and 34 PCV2 sequences originating from China and other countries. The tree was constructed using the Maximum Likelihood algorithm. The 34 reference strains which are representatives of all PCV2 genotypes are marked with a black circle
Phylogenetic analysis of the complete genome showed that current PCV2 strains in this study could be divided into PCV2a (1/95), PCV2b (39/95), PCV2d (43/95), PCV2e (10/95) and PCV2h (2/95), as shown in Fig. 2. The genotype PCV2b was further divided into PCV2b-1A (5/95) and PCV2b-1B (34/95). All 95 strains with a complete genome phylogeny have the same classification with respect to the ORF2-based phylogeny, except for two strains (GXNN0301and GXNN0604) which were clustered to PCV2g (Additional file 1: Figure S1). Among the 5 sub-genotypes, PCV2b was dominant in the porcine population from 2002 to 2008. The newly identified sub-genotype, PCV2d, was found from 2003 and its presence has increased year by year. PCV2b and PCV2d are two predominant genetic groups which circulated in the Guangxi Province between 2009 and 2013 and PCV2d is the predominant genotype circulating in the swine population of this region since 2014 (Fig. 3).

The alignment of Cap for PCV2. A multiple alignment of PCV2 Cap was performed by Clustal W. The grey areas show the antibody recognition domains and the immune-dominant decoy epitope described previously [30, 31]. The boxes show the motifs of PCV2a, PCV2b and PCV2d which have been previously described [13, 28, 29]

The time distribution and genotype of the 95 PCV2 strains in this study
Discussion
In our previous study, 181 of the 371 (48.8%) samples collected were positive for PCV2, indicating that PCV2 is widely distributed among swine populations in Guangxi, China. There is extensive genetic variability in four major regions at amino acid positions 53–90, 121–136, 169–218 and 232–234. There are critical aa’s within some signature motifs which are reported to be important for differentiation of the PCV2 genotype [13, 28, 29] as well as regions such as antibody epitopes, immune-dominant decoy epitopes and key aa’s which determine virulence [30,31,32,33,34] which were found in the Cap protein domains of some strains. ORF2 is the major structural protein of PCV2 that is believed to be involved in diverse functions such as receptor binding, host immune response and viral replication [6, 32,33,34]. Therefore, a small number of mutations might result in antigenic variations or increased pathogenicity of the virus.
Full length genome and ORF2 based phylogenetic trees showed all these strains are present in 5 sub-genotypes (PCV2a, PCV2b, PCV2d, PCV2e and PCV2h). Global genetic analysis indicated that the PCV2 evolution trace was PCV2a to PCV2b to PCV2d [13]. Many previous studies showed that genotype shift from PCV2a to PCV2b occurred in 2002 in mainland China and PCV2b has been the predominant genotype since then [19, 23, 24]. A similar major shift from PCV2a to PCV2b has also occurred in many countries on a global scale prior to 2003 [35, 36]. Consistent with these previous studies, our study shows that only one strain (PCV2a) was found in 2016, suggesting a major shift from PCV2a to PCV2b had occurred in Guangxi Province in or prior to 2002. The PCV2b is the predominant genotype found between 2003 and 2011.
Many studies have indicated that the rapid genotype shift from PCV2a to PCV2b was related to the appearance of PMWS cases at the farmyard level together with an accompanying increase in clinical severity [37,38,39]. However, there are no significant difference in virulence between PCV2a and PCV2b-inoculated groups under experimental conditions [40, 41]. In this study, we also showed there was no significant relationship between the infection caused by PCV2b and PMWS cases (data not shown). Both PCV2a and PCV2b could be detected in the healthy pigs and in PWMS-affected pigs (Table 1). Our results showed that PCV2b and PCV2d were the two predominant genetic groups circulating in southern China between 2008 and 2013. PCV2d was the predominant genotype seen since 2014, indicating that the process leading to the genotype shift from PCV2b to PCV2d had already begun at the province-wide scale in these subsequent years.
In 2010, a variant PCV2 mutant strain designated as mPCV2b, and now classified as PCV2d, was identified and the prevalence rate of mPCV2b appears to have increased both in China and the USA. In this study, a dramatic increase in detection of the PCV2d variant has been seen since 2014. Whether the PCV2d is more pathogenic in pigs is controversial. One study conducted by Guo et al. showed that mPCV2, now classified as PCV2d, induced more severe clinical, pathological, and virological manifestations than the genotypes PCV2a and PCV2b in conventional pigs [23]. However, another study showed that there was no significant difference in pathogenicity between PCV2a/b and mPCV2 in caesarean-derived, colostrum-deprived pigs [42].
In this study, we showed there was no significant relationship between the predominance of PCV2d and PMWS cases. Therefore, the pathogenicity of mPCV2 in pigs and the association between increased mPCV2 prevalence and its clinical manifestation in the field needs to be further studied.
Conclusions
This study reveals the complex genetic diversity of PCV2 and improves our understanding regarding the epidemiological trends of PCV2 sub-genotypes in China.
Methods
Sample collection, viral DNA extraction and PCV2 detection
Field samples (sera, lungs, lymph nodes and spleens) from commercial pig farms in different regions of Guangxi province between 2002 and 2016 were submitted to Laboratory of Animal infectious Diseases and Molecular Immunology, Guangxi University, Nanning for PCV2 testing. Total viral DNA was extracted directly from sera and tissue samples using Virus Genome Extract DNA kit according to the manufacturer’s instructions (TIANGEN, Inc., Beijing, China). Viral DNAs were eluted in 50 μL of ddH2O and were stored at − 30 °C until used. All the samples were screened for PCV2 by PCR using primers (5′-CCGCGGGCTGGCTGAACTT-3′) and (5′-ACCCCCGCCACCGCTACC -3′). Thermal cycling conditions were 94 °C for 3 min, followed by 35 cycles of 94 °C for 40 s, 60 °C for 40s, 72 °C for 50 s, and a final elongation step at 72 °C for 10 min. Finally, the PCR products were analyzed using 1.0% agarose gel electrophoresis ultraviolet imaging. Positive samples were determined with 1154 bp amplified products. Positive amplicons were purified using E.Z.N.A.TM Gel Extraction Kit (OMEGA, USA) and were further cloned into pBST-IIvector (TIANGEN, Inc., Beijing, China) for nucleotide sequencing by using primer T7 or T3 (HuaDa Gene, Inc., China).
PCV2 amplification and sequence determination
PCV2 positive samples were used for full-length genome amplification and sequencing. The forward primer (5′ GAACCGCGGGCTGGCTGAACTTTTGAAAGT 3′) and reverse primer (5′ GCACCGCGGAAATTTCTGACAAACGTTACA 3′) were used for amplification of the full genome. PCR reaction conditions were 94 °C for 5 min, followed by 30 cycles of 94 °C for 1 min, 55 °C for 1 min, 72 °C for 2 min, and a final elongation step at 72 °C for 10 min. The PCR products were purified with E.Z.N.A.TM Gel Extraction Kit (OMEGA, USA) and cloned into a pBST-IIvector (TIANGEN, Inc., Beijing, China). Positive clones were sequenced in both directions using universal primers T7 and SP6 promoter-specific primers (HuaDa Gene, Inc., China).
Phylogenetic tree analysis
Differences of the amino acid sequences derived from the ORF2 gene of the 95 strains and representative isolates from China and other countries were analyzed and aligned using DNAstar software (DNASTAR Inc., Madison, WI, USA). MEGA version 6.0 was used to evaluate phylogenetic relationships by the Maximum Likelihood method with 1000 bootstrap replicates. A Maximum Likelihood phylogenetic tree was constructed including the 95 different complete genomes or ORF2 genes from this study and the complete genome or ORF2 gene sequences of the 34 representative isolates from China and other countries representative of all PCV2 genotypes, containing the considered PCV2 genotypes a, b, c, d, e, f, g and h. The sequences obtained in this study were submitted to the GenBank database under the accession numbers (MH465398~MH465491 and MH481748).
Statistical analysis
PASW Statistics 18 software (PASW, Inc., an IBM Company, Chicago, IL) was used to perform χ2 test to evaluate the association of PCV2b and PCV2d with PMWS cases. P values of < 0.05 were considered statistically significant.
Abbreviations
- CT:
-
Congenital tremors type II
- ORFs:
-
Open reading frames
- PCV2:
-
Porcine circovirus type 2
- PCVAD:
-
PCV2-associated diseases
- PDNS:
-
Porcine dermatitis and nephropathy syndrome
- PMWS:
-
Post-weaning multi-systemic wasting syndrome
- PRDC:
-
Porcine respiratory disease complex
References
Allan GM, Mc Neilly F, Meehan BM, Kennedy S, Mackie DP, Ellis JA, Clark EG, Espuna E, Saubi N, Riera P, et al. Isolation and characterisation of circoviruses from pigs with wasting syndromes in Spain, Denmark and Northern Ireland. Vet Microbiol. 1999;66(2):115–23.
Allan GM, McNeilly E, Kennedy S, Meehan B, Moffett D, Malone F, Ellis J, Krakowka S. PCV-2-associated PDNS in Northern Ireland in 1990. Porcine dermatitis and nephropathy syndrome. Vet Rec. 2000;146(24):711–2.
Ellis J, Hassard L, Clark E, Harding J, Allan G, Willson P, Strokappe J, Martin K, McNeilly F, Meehan B, et al. Isolation of circovirus from lesions of pigs with postweaning multisystemic wasting syndrome. Can Vet J. 1998;39(1):44–51.
Rosell C, Segales J, Ramos-Vara JA, Folch JM, Rodriguez-Arrioja GM, Duran CO, Balasch M, Plana-Duran J, Domingo M. Identification of porcine circovirus in tissues of pigs with porcine dermatitis and nephropathy syndrome. Vet Rec. 2000;146(2):40–3.
Hamel AL, Lin LL, Nayar GP. Nucleotide sequence of porcine circovirus associated with postweaning multisystemic wasting syndrome in pigs. J Virol. 1998;72(6):5262–7.
Nawagitgul P, Morozov I, Bolin SR, Harms PA, Sorden SD, Paul PS. Open reading frame 2 of porcine circovirus type 2 encodes a major capsid protein. J Gen Virol. 2000;81(Pt 9:2281–7.
Liu J, Chen I, Kwang J. Characterization of a previously unidentified viral protein in porcine circovirus type 2-infected cells and its role in virus-induced apoptosis. J Virol. 2005;79(13):8262–74.
He J, Cao J, Zhou N, Jin Y, Wu J, Zhou J. Identification and functional analysis of the novel ORF4 protein encoded by porcine circovirus type 2. J Virol. 2013;87(3):1420–9.
Lv Q, Guo K, Xu H, Wang T, Zhang Y. Identification of putative ORF5 protein of porcine circovirus type 2 and functional analysis of GFP-fused ORF5 protein. PLoS One. 2015;10(6):e0127859.
Choi CY, Choi YC, Park IB, Lee CH, Kang SJ, Chun T. The ORF5 protein of porcine circovirus type 2 enhances viral replication by dampening type I interferon expression in porcine epithelial cells. Vet Microbiol. 2018;226:50–8.
Cheung AK. The essential and nonessential transcription units for viral protein synthesis and DNA replication of porcine circovirus type 2. Virology. 2003;313(2):452–9.
Olvera A, Cortey M, Segales J. Molecular evolution of porcine circovirus type 2 genomes: phylogeny and clonality. Virology. 2007;357(2):175–85.
Xiao CT, Halbur PG, Opriessnig T. Global molecular genetic analysis of porcine circovirus type 2 (PCV2) sequences confirms the presence of four main PCV2 genotypes and reveals a rapid increase of PCV2d. J Gen Virol. 2015;96(Pt 7):1830–41.
Cai L, Ni J, Xia Y, Zi Z, Ning K, Qiu P, Li X, Wang B, Liu Q, Hu D, et al. Identification of an emerging recombinant cluster in porcine circovirus type 2. Virus Res. 2012;165(1):95–102.
Franzo G, Segales J. Porcine circovirus 2 (PCV-2) genotype update and proposal of a new genotyping methodology. PLoS One. 2018;13(12):e0208585.
Grau-Roma L, Crisci E, Sibila M, Lopez-Soria S, Nofrarias M, Cortey M, Fraile L, Olvera A, Segales J. A proposal on porcine circovirus type 2 (PCV2) genotype definition and their relation with postweaning multisystemic wasting syndrome (PMWS) occurrence. Vet Microbiol. 2008;128(1–2):23–35.
Cortey M, Olvera A, Grau-Roma L, Segales J. Further comments on porcine circovirus type 2 (PCV2) genotype definition and nomenclature. Vet Microbiol. 2011;149(3–4):522–3.
Franzo G, Cortey M, de Castro AM, Piovezan U, Szabo MP, Drigo M, Segales J, Richtzenhain LJ. Genetic characterisation of porcine circovirus type 2 (PCV2) strains from feral pigs in the Brazilian Pantanal: An opportunity to reconstruct the history of PCV2 evolution. Vet Microbiol. 2015;178(1–2):158–62.
Wang F, Guo X, Ge X, Wang Z, Chen Y, Cha Z, Yang H. Genetic variation analysis of Chinese strains of porcine circovirus type 2. Virus Res. 2009;145(1):151–6.
Jantafong T, Boonsoongnern A, Poolperm P, Urairong K, Lekcharoensuk C, Lekcharoensuk P. Genetic characterization of porcine circovirus type 2 in piglets from PMWS-affected and -negative farms in Thailand. Virol J. 2011;8:88.
Harmon KM, Gauger PC, Zhang J, Pineyro PE, Dunn DD, Chriswell AJ. Whole-genome sequences of novel porcine circovirus type 2 viruses detected in swine from Mexico and the United States. Genome Announc. 2015;3(6):e01315–15.
Davies B, Wang X, Dvorak CM, Marthaler D, Murtaugh MP. Diagnostic phylogenetics reveals a new porcine circovirus 2 cluster. Virus Res. 2016;217:32–7.
Guo LJ, Lu YH, Wei YW, Huang LP, Liu CM. Porcine circovirus type 2 (PCV2): genetic variation and newly emerging genotypes in China. Virol J. 2010;7:273.
Wang H, Gu J, Xing G, Qiu X, An S, Wang Y, Zhang C, Liu C, Gong W, Tu C, et al. Genetic diversity of porcine circovirus type 2 in China between 1999-2017. Transbound Emerg Dis. 2019;66(1):599–605.
Yang S, Yin S, Shang Y, Liu B, Yuan L, Zafar Khan MU, Liu X, Cai J. Phylogenetic and genetic variation analyses of porcine circovirus type 2 isolated from China. Transbound Emerg Dis. 2018;65(2):e383–92.
Xiao CT, Halbur PG, Opriessnig T. Complete genome sequence of a novel porcine circovirus type 2b variant present in cases of vaccine failures in the United States. J Virol. 2012;86(22):12469.
Jiang CG, Wang G, Tu YB, Liu YG, Wang SJ, Cai XH, An TQ. Genetic analysis of porcine circovirus type 2 in China. Arch Virol. 2017;162(9):2715–26.
Cheung AK, Lager KM, Kohutyuk OI, Vincent AL, Henry SC, Baker RB, Rowland RR, Dunham AG. Detection of two porcine circovirus type 2 genotypic groups in United States swine herds. Arch Virol. 2007;152(5):1035–44.
Cheung AK. Homologous recombination within the capsid gene of porcine circovirus type 2 subgroup viruses via natural co-infection. Arch Virol. 2009;154(3):531–4.
Trible BR, Rowland RR. Genetic variation of porcine circovirus type 2 (PCV2) and its relevance to vaccination, pathogenesis and diagnosis. Virus Res. 2012;164(1–2):68–77.
Trible BR, Kerrigan M, Crossland N, Potter M, Faaberg K, Hesse R, Rowland RR. Antibody recognition of porcine circovirus type 2 capsid protein epitopes after vaccination, infection, and disease. Clin Vaccine Immunol. 2011;18(5):749–57.
Allemandou A, Grasland B, Hernandez-Nignol AC, Keranflec'h A, Cariolet R, Jestin A. Modification of PCV-2 virulence by substitution of the genogroup motif of the capsid protein. Vet Res. 2011;42:54.
Shang SB, Jin YL, Jiang XT, Zhou JY, Zhang X, Xing G, He JL, Yan Y. Fine mapping of antigenic epitopes on capsid proteins of porcine circovirus, and antigenic phenotype of porcine circovirus type 2. Mol Immunol. 2009;46(3):327–34.
Fenaux M, Opriessnig T, Halbur PG, Elvinger F, Meng XJ. Two amino acid mutations in the capsid protein of type 2 porcine circovirus (PCV2) enhanced PCV2 replication in vitro and attenuated the virus in vivo. J Virol. 2004;78(24):13440–6.
Rose N, Opriessnig T, Grasland B, Jestin A. Epidemiology and transmission of porcine circovirus type 2 (PCV2). Virus Res. 2012;164(1–2):78–89.
Dupont K, Nielsen EO, Baekbo P, Larsen LE. Genomic analysis of PCV2 isolates from Danish archives and a current PMWS case-control study supports a shift in genotypes with time. Vet Microbiol. 2008;128(1–2):56–64.
Cortey M, Pileri E, Sibila M, Pujols J, Balasch M, Plana J, Segales J. Genotypic shift of porcine circovirus type 2 from PCV-2a to PCV-2b in Spain from 1985 to 2008. Vet J. 2011;187(3):363–8.
Gagnon CA, Tremblay D, Tijssen P, Venne MH, Houde A, Elahi SM. The emergence of porcine circovirus 2b genotype (PCV-2b) in swine in Canada. Can Vet J. 2007;48(8):811–9.
Carman S, Cai HY, DeLay J, Youssef SA, McEwen BJ, Gagnon CA, Tremblay D, Hazlett M, Lusis P, Fairles J, et al. The emergence of a new strain of porcine circovirus-2 in Ontario and Quebec swine and its association with severe porcine circovirus associated disease--2004-2006. Can J Vet Res. 2008;72(3):259–68.
Gauger PC, Lager KM, Vincent AL, Opriessnig T, Kehrli ME Jr, Cheung AK. Postweaning multisystemic wasting syndrome produced in gnotobiotic pigs following exposure to various amounts of porcine circovirus type 2a or type 2b. Vet Microbiol. 2011;153(3–4):229–39.
Opriessnig T, Ramamoorthy S, Madson DM, Patterson AR, Pal N, Carman S, Meng XJ, Halbur PG. Differences in virulence among porcine circovirus type 2 isolates are unrelated to cluster type 2a or 2b and prior infection provides heterologous protection. J Gen Virol. 2008;89(Pt 10):2482–91.
Opriessnig T, Xiao CT, Gerber PF, Halbur PG, Matzinger SR, Meng XJ. Mutant USA strain of porcine circovirus type 2 (mPCV2) exhibits similar virulence to the classical PCV2a and PCV2b strains in caesarean-derived, colostrum-deprived pigs. J Gen Virol. 2014;95(Pt 11:2495–503.
Acknowledgements
The authors would like to thank Dr. Dev Sooranna, Imperial College London, for editing the manuscript.
Funding
This study was supported by National Key Research and Development Program of China (2018YFD0500100), Key Projects of Guangxi Natural Science Foundation (2018GXNSFDA281021) and Guangxi Science and Technology Bureau (No. AA17204057). They provided the funds as well as monitoring and evaluation of the project.
Availability of data and materials
All relevant data are within this paper. The data analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
YJ and QY conducted experiment, analyzed the data. YZ assisted sample preparation and experiment. CY and KO shared ideas and discussed the research data. HW and WZ contributed to supervision, had the idea for the project, and directed the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
For all porcine clinical samples used in this study, written consents were obtained from farm owners and all procedures were carried out in strict accordance with the Animal Ethics Procedures and Guidelines of the People’s Republic of China. All the animal protocols in this study were approved by the Ethics Committee of Guangxi University.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Phylogenetic tree based on PCV2 ORF2 sequences. Phylogenetic tree based on a comparison of 129 PCV2 ORF2 sequences, including the 95 sequences from this study and 34 PCV2 sequences originating from China and other countries. The tree was constructed using the Maximum Likelihood algorithm. The 34 reference strains which are representatives of all PCV2 genotypes are marked with a black circle. (PPTX 203 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yao, J., Qin, Y., Zeng, Y. et al. Genetic analysis of porcine circovirus type 2 (PCV2) strains between 2002 and 2016 reveals PCV2 mutant predominating in porcine population in Guangxi, China. BMC Vet Res 15, 118 (2019). https://doi.org/10.1186/s12917-019-1859-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-019-1859-z