
Abstract
Background
Muscovy duck parvovirus (MDPV) causes high mortality and morbidity in Muscovy ducks, with the pathogenesis of the virus still unknown in many respects. Specific MDPV detection is often rife with false positive results because of high identity at the genomic nucleotide level and antigenic similarity with goose parvovirus (GPV). The objective of this study was to develop a sensitive, highly specific, and repeatable TaqMan-based real-time PCR (qPCR) assay for facilitating the molecular detection of MDPV.
Results
The specific primers and probe were designed based on the conserved regions within MDPVs, but there was a variation in GPVs of the nonstructural (NS) genes after genetic comparison. After the optimization of qPCR conditions, the detection limit of this qPCR assay was 29.7 copies/μl. The assay was highly specific for the detection of MDPV, and no cross-reactivity was observed with other non-targeted duck-derived pathogens. Intra- and inter-assay variability was less than 2.21%, means a high degree of repeatability. The diagnostic applicability of the qPCR assay was proven that MDPV-positive can be found in cloacal swabs samples, Muscovy duck embryos and newly hatched Muscovy ducklings.
Conclusions
Our data provided incidents that MDPV could be possible vertically transmitted from breeder Muscovy ducks to Muscovy ducklings. The developed qPCR assay in the study could be a reliable and specific tool for epidemiological surveillance and pathogenesis studies of MDPV.
Similar content being viewed by others
Background
The family Parvoviridae is comprised of two subfamily members, Densovirinae and Parvovirinae. The subfamily Parvovirinae contains eight distinct genera: Amdoparvovirus, Aveparvovirus, Bocaparvovirus, Copiparvovirus, Dependoparvovirus, Erythroparvovirus, Protoparvovirus, and Tetraparvovirus. At present, the International Committee on Taxonomy of Viruses (ICTV) has classified Muscovy duck parvovirus (MDPV) and goose parvovirus (GPV) as a single species (namely, Anseriform dependoparvovirus 1), which is classified into the genus Dependoparvovirus in subfamily Parvovirinae due to similar genetic properties and evolutionary origins (https://talk.ictvonline.org/taxonomy/).
Reportedly, the genomes of MDPVs and GPVs contain a single copy of the linear, single-stranded DNA genome of approximately 5100 nucleotides in length. The genomes of these viruses are flanked by identical inverted terminal repeats (ITR) at both the 5′- and 3′-terminus. The ITR can fold on itself to form a palindromic hairpin structure. A terminal resolving site (TRS), Rep protein binding site (RBS), and transcription factor binding sites can be found in ITR, which were involved in viral replication, packaging and transcription. There are two major open reading frames (ORFs) in both MDPVs and GPVs genome. The left ORF that encodes for the nonstructural (NS) protein, which is involved in viral replication and regulatory function. However, the right ORF that produces three capsid proteins (VP1, VP2 and VP3), which plays important roles in virus tropism, host range, and pathogenicity. In addition, VP2 and VP3 contain the same carboxyl-terminal portion of VP1 of the viruses, which was generated by differential alternative splicing of mRNA [1,2,3,4,5,6,7,8].
Normally, GPVs have been found in goslings, Muscovy ducklings [5, 8], swans [9], and Anser cygnoides [10], whereas MDPV has only been discovered in Muscovy ducklings. MDPV infection was initially described by Professor Lin in our laboratory in the early 1980s [11]. Muscovy ducklings infected by MDPV were characterized by watery diarrhea, wheezing, and locomotor dysfunction. MDPV is mainly observed in Muscovy ducklings less than three-week-old, with the mortality rate reaching as high as 80% depending on age [3, 7, 11].Then the disease was widespread in China, leading huge economic loss to waterfowl husbandry due to the high mortality and morbidity.
A previous study revealed that MDPV and GPV are also pathogenic to Muscovy ducklings, even in the same Muscovy duck flocks [8, 12]. Compared with the GPV virulent strain B and MDPV virulent strain FM, they share more than 80.0% nucleotide similarity at the genome level. In addition, these two viruses exhibit nucleotides and amino acids identities of 83.0 and 90.6% at NS gene level, and 81.5% and 87.6% at VP1 gene level, respectively [1,2,3]. The high identity at the amino acids level of the VP1 protein indicates potential immunogenic cross-reactivity between MDPVs and GPVs [13,14,15]. Therefore, differentiating between MDPV and GPV in Muscovy ducklings is essential. Nevertheless, the high homologies in nucleotide identities and immunogenic cross-reactivity between MDPVs and GPVs, increases the risk of omissive and mistaken diagnoses for the specific detection of MDPV.
Recently, a TaqMan-based real-time quantitative PCR (qPCR) assays have gained wide acceptance due to their rapid nature, sensitivity, reproducibility, and the reduced risk of carry-over contamination as a result of the specific TaqMan probe, which had been widely used for viral epidemiological surveillance and pathogenesis studies [16,17,18,19]. Thereby, the aim of this study was to design a fully validated, reliable, and highly specific TaqMan-based real-time quantitative PCR assay for precise detection of MDPV infection based on specific primers and probe, designed by targeting the conserved region of the MDPV NS gene after bioinformatics analysis.
Methods
Viruses and bacteria strains
Avian influenza virus (H9N2 AIV), avian Tembusu virus (ATmV), duck hepatitis virus type 1 and 3 (DHAV-1 and DHAV-3), Muscovy duck reovirus (MDRV), duck adenovirus A (DAdV-A), duck enteritis virus (DEV), Muscovy duck origin goose parvovirus (GPV), novel goose parvovirus (N-GPV), Escherichia coli (E. coli.), Pasteurellamultocida (P.M.), Rimerella anatipstifer (R.A.) and Salmonella spp. (S.S.) were isolated and kept in our laboratory, which was decribed the same as previously reported [12, 20, 21].
Nucleic acids extraction and cDNAs preparation
Viral RNAs (RNA viruses, i.e. AIV, ATmV, DHAV-1 and DHAV-3, MDRV) were extracted using EasyPure Viral DNA/RNA Kit (Tiangen, Beijing, China), viral DNAs (DNA viruses, i.e. DAdV-A, DEV, GPV and N-GPV) were extracted using EasyPure Micro Genomic DNA Kit (Transgen Bioteck, Beijing, China), according to instructions provided by the manufacturer. cDNAs (AIV, ATmV, DHAV-1 and DHAV-3, MDRV) were prepared with viral RNAs (with approximate 100 ng of viral RNA) using TransScript II All-in-One First-Strand cDNA Synthesis SuperMix (One-Step gDNA Removal) (Tiangen, Beijing, China). Bacteria genomic DNAs (bacteria pathogens, i.e. E. coli., P.M., R.A. and S.S.) were extracted using EasyPure Bacteria Genomic DNA Kit (Transgen, Beijing, China). DNAs and cDNAs were then quantified using a NANODROP 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). All extracted DNAs and cDNAs templates were stored at − 80 °C until use.
Primers and probe selection and design
Primer and probe selection was performed on the evolutionarily most conserved regions of the NS gene of MDPV. Briefly, a total of 15 MDPVs and 37 GPVs NS gene sequences were downloaded from the GenBank database (https://www.ncbi.nlm.nih.gov/nucleotide/), and these 52 NS gene sequences were aligned using the Lasergene package MegAlign program by ClustalW method. The identification of the conserved region, which was highly conserved in MDPVs, but there was a characteristics variation in GPVs. The obvious different region between MDPVs with GPVs, was selected for the primers and TaqMan probe design. The forward primer MDPV-qF (5’-TACGAATGAACAAACCAA-3′), the reverse primer MDPV-qR (5′- CGCTCTTAATATCTCCTCTA-3′), and the TaqMan probe MDPV-qP (FAM-5′- TGAACGAGCGAATGAGCCTTCC-3′-Eclipse) were designed using Primer Premier Software version 5.0 (Premier Biosoft, Palo Alto, CA, USA). The length of amplicon was 118 base pairs (bp). Primers (MDPV-qF and MDPV-qR) and probe (MDPV-qP) were verified by Basic Local Alignment Search Tool (BLAST, https://blast.ncbi.nlm.nih.gov/Blast.cgi) for specificity analysis, then these verified primers and probe were synthesized by a commercial company (TaKaRa, Dalian, China).
Construction of recombinant plasmid containing the NS gene of MDPV
The NS gene (1884 bp) of MDPV (strain FJM5) [8] was amplified by PCR, with the primer sets of forward primer (NSF) 5’-ATGGCATTTTCTAGGCCTCTTCA-3′ and reverse primer (NSR) 5’-TTATTGTTCATTCTCCATATCAT-3′. A conventional PCR was performed in a Thermal Cycler Dice (TaKaRa, OTSU, SHIGA, Japan), the isolated DNAs were used as template in a reaction volume of 50 μl reaction mixture containing 25 μl ThermoScientific DreamTaq Green PCR Master Mix (2×) (Thermo Fisher Scientific Inc., Shanghai, China), 1 μl primers NSF and NSR (20 μM each), 1 μl DNA template, and 22 μl Nuclease-free water. The PCR reaction was conducted using a program that included with an initial denaturation at 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 50 s, annealing at 53 °C for 35 s, elongation at 72 °C for 120 s, and then a final extension at 72 °C for 10 min.
The amplified PCR products were then subjected to electrophoresis on 1.0% agarose gels for analysis. The expected PCR amplicons were purified and then T-A cloned using the pMD18-T Vector Cloning Kit (TaKaRa, Dalian, China). Then the transformants were identified by a commercial company (Sangon, Shanghai, China) for nucleotide sequencing. The selected recombinant plasmid, pMD18-NS, was used as the standard plasmid. The plasmid pMD18-NS was then quantified using a NANODROP 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). According to the formula described by Yun et al. [22], the copy numbers of the plasmid pMD18-NS was calculated. Serial 10-fold dilutions of plasmid pMD18-NS, were diluted using EASY Dilution (TaKaRa, Dalian, China). All the serial diluted plasmids (ranging from 2.97 × 107 to 2.97 × 100 copies/μl), were stored at − 80 °C until use.
Real-time PCR assay
Real-time PCR amplification and detection were carried out on Mastercycler ep realplex (Eppendorf, Germany). The concentration of the primers, probe, and templates were optimized based on the obtained fluorescence and lowest threshold cycle (Ct). The optimized TaqMan-based PCR was prepared in a final volume of 25 μl containing 1 μl DNA template, 12.5 μl Premix Ex Taq (Probe qPCR, TaKaRa, Dalian, China), 0.5 μl of each primer (MDPV-qF and MDPV-qR, 10 μmol/l each), 2 μl probe (MDPV-qP, 5 μmol/l), and 8.5 μl Nuclease-free water to to adjust the reaction volume (total reaction volume of 25 μl). The mixed reactions was conducted in a single tube in a Mastercycler ep realplex by using the following thermoprofile: 40 cycles of 95 °C for 5 s, 58 °C for 10 s, and 72 °C for 15 s. 10-fold serial dilutions of plasmid pMD18-NS, containing different copy numbers of DNA (2.97 × 106 to 2.97 × 101 copies/μl) were conducted to generate the standard curve. All of the reactions were conducted in triplicate simultaneously. Analysis of each assay was conducted with CalQplex software (Mastercycler ep realplex, Eppendorf, Germany) according to the instruction manual. The software automatically uses the Ct values of serial dilutions of standards to calculate a standard curve, which shows the Ct values as a function of the amount of different copy numbers of DNA.
Sensitivity analysis
To evaluate the limit of detection (LOD) of the qPCR assay, 10-fold serial dilutions of plasmid DNA standard (ranging from 2.97 × 105 to 2.97 × 100 copies/μl) were prepared to determine the sensitivity. Each concentration was run in triplicate. Meanwhile, conventional PCR (cPCR) was performed with primers (NSF1 and NSR1) (NSF1, 5′- CAATGGGCTTTTACCAATATGC-3′ and NSR1, 5′- ATTTTTCCCTCCTCCCACCA-3′) and the same standard plasmid, in order to determine the LOD of cPCR assay. The cPCR reaction mixtures and thermal profile was described the same as previously reported [12]. PCR products were visualized following electrophoresis of 5 μl of each reaction in a 1.0% agarose gel according to instructions provided by the manufacturer. The LOD between the cPCR and qPCR assay were then compared.
Specificity analysis and reproducibility analysis
To determine the specificity of the qPCR assay, ten ng of extracted DNAs and cDNAs templates were used for the specificity analysis. The qPCR assay was carried out in triplicate to amplify a panel of duck-derived pathogens, i.e. H9N2 AIV, ATmV, DHAV-1, DHAV-3, MDRV, DAdV-A, DEV, GPV, N-GPV, E. coli., P.M., R.A. and S.S.. Nuclease-free water also used in the qPCR run to validate the specificity of qPCR assay as negative control. For reproducibility analysis of the qPCR assay, the 10-fold dilutions of pMD18-NS (concentration with 2.97 × 105, 2.97 × 103, and 2.97× 101 copies/μl) were tested to evaluate the coefficient of variation (CV). For intra-assay variability of qPCR assay, triplicates of each dilution were analyzed, and the CVs were calculated according to the formula of the geometric mean Ct values deviation. For inter-assay variability, a coefficient of variation which expresses the standardized measure of dispersion with different time.
Clinical samples detection of MDPV
Detection of MDPV in field samples
In order to validate the qPCR assay, a total of 75 individuals Muscovy duckling-origin cloacal swabs samples (less than three-week) with diarrheal symptoms were collected in Fujian, Jiangxi, Guangdong, Jiangsu, and Zhejiang provinces, China. All of the suspensions were subjected to three freeze-thaw cycles and then centrifuged at 8000 rpm at 4 °C for 30 min. Viral DNAs were extracted from the harvested supernatants using EasyPure Micro Genomic DNA Kit (Transgen Bioteck, Beijing, China). Conventional PCR was preformed simultaneously.
Detection of MDPV in Muscovy duck embryos and newly hatched ducklings
Previous study indicates possible vertical transmission of N-GPV and suggests that N-GPV may be transmitted from breeder ducks to ducklings in ovo [23]. In order to determine the hypothesis whether MDPV could be possible vertically transmitted or not, 20 Muscovy duck embryos (15-day post fertilization) and 20 newly hatched Muscovy ducklings (1-day-old) were collected from the diseased farms where the virus (FJM5 strain) was discovered [8]. The liver of each embryo and newly hatched duckling was pooled and regarded as one sample. These samples were homogenized in phosphate-buffered saline (PBS) (20%, w/v). Viral DNAs were extracted from tissue homogenates of liver using EasyPure Micro Genomic DNA Kit (Transgen Bioteck, Beijing, China). Conventional PCR was preformed simultaneously. All of the cPCR-positive amplicons were harvested, T-A cloned, and then sequenced to verify the results for Sanger seuquencing at Sangon (Shanghai, China) in both directions.
Results
NS gene analysis
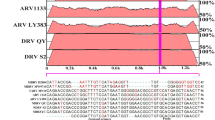
We compared a total of 52 NS gene sequences (including 15 MDPVs and 37 GPVs) downloaded from the GenBank database. We found that within the MDPV cluster, there was a higher nucleotide identity (more or equal than 98.0%) than within the GPV cluster (more or equal than 93.3%). In addition, the NS gene homology between GPV cluster and MDPV cluster ranged from 80.8 to 83.4% (Table 1). The primers MDPV-qF and MDPV-qR, and the TaqMan probe MDPV-qP variation within MDPVs are listed in Table 2. Moreover, the 3′-terminal of MDPV-qP at position 1521–1524 was TTCC, while the sequence at this position in the 37 GPVs was either GGAG (54.05%, 20/37), AGAG (40.54%, 15/37), AGAT (2.70%, 1/37), or GAAG (2.70%, 1/37). This data demonstrated that sequences at positions 1521–1524 were significantly different between MDPVs and GPVs, which can be chosen for specific probe design.
Real-time PCR
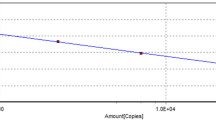
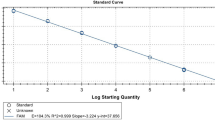
The generated standard curve showed linearity over the 2.97 × 106 to 2.97 × 101 copies/μl range, with a slope of − 3.358 and the Y-intercept was 34.93, efficiency of 0.99, which was conducted by software of Mastercycler ep realplex (Eppendorf, Germany) (Fig. 1). Using the viral DNAs, cDNAs, bacterial DNAs and Nuclease-free water, the results showed that only MDPV was detected by the TaqMan-based real-time PCR assay. No fluorescence signal was detected in control samples of H9N2 AIV, ATmV, DHAV-1, DHAV-3, MDRV, DAdV-A, DEV, GPV, N-GPV, E. coli., P.M., R.A., S.S. and Nuclease-free water (Fig. 2), which indicated the excellent specificity of the qPCR assay. The LOD of the qPCR assay was 29.7 copies/μl (Fig. 3a). In contrast, conventional PCR had a detection limit of 2.97 × 102 copies/μl (Fig. 3b). The intra-assay CVs of pMD18-NS (concentration with 2.97 × 105, 2.97 × 103, and 2.97× 101 copies/μl) were 0.59%, 0.44% and 1.43%. The inter-assay CVs were 0.66%, 0.63% and 2.21% (listed in Table 3), which indicate a high degree of repeatability.

Standard curve of TaqMan-based real-time PCR assay

Specificity of TaqMan-based real-time PCR assay. 1: MDPV; Controls: H9N2 AIV, ATmV, DHAV-1, DHAV-3, MDRV, DAdV-A, DEV, GPV, N-GPV, E. coli., P.M., R.A., S.S. and Nuclease-free water. These controls were all found with no fluorescence signal

a Sensitivity of TaqMan-based real-time PCR assay for MDPV detection. b Sensitivity of conventional PCR assay for MDPV detection. 1–6: a serial of ten-fold dilutions plasmid DNA (2.97 × 105 to 2.97 × 100 copies/μl); 7: negative control; M: DL2000 DNA Marker
Clinical samples application
Senventy-five cloacal swabs from Muscovy ducklings with diarrhea were evaluated of MDPV diagnosis by the TaqMan-based real-time PCR and conventional PCR assays. As summarized in Table 4, for 75 cloacal swabs evaluated samples, 10 (13.33%) were MDPV-positive by qPCR assay. However, cPCR results showed 7 were MDPV-positive. For embryonic samples, 4 of 20 (20.0%) were MDPV-positive by qPCR assay and 2 of 20 (10.0%) were MDPV-positive by cPCR assay. For newly hatched ducklings, 5 of 20 (25.0%) were MDPV-positive by qPCR assay and 4 of 20 (20.0%) were MDPV-positive by cPCR assay, respectively. The samples tested with MDPV-positive by cPCR assay, were also tested MDPV-positive by qPCR assay. The viral DNA copy numbers in the positive samples when detecting MDPV DNAs were listed in Table 4. These findings provide evidence of possible vertical transmission of MDPV.
Sequences analysis
A total of 13 (7 from cloacal swabs, 2 from embryonic samples, and 4 from newly hatched ducklings) amplicons from cPCR positive were harvested, purified, T-A cloned and then sequenced. The cloned sequences shared ≥99.1% nucleotide identity with MDPV (strain FM). Moreover, the cloned sequences from cPCR positive samples could specifically (100%) match the primers (MDPV-qF and MDPV-qR) and probe (MDPV-qP) listed in Table 2.
Discussion
The current methods for the detection of MDPV, such as virus isolation, immunological-based assays, and electron microscopy, have proven to be laborious and time-consuming. Conventional PCR technology has been used for differentiation between MDPV and GPV, and the process includes restriction enzyme digestion, agarose gel electrophoresis, and DNA sequencing [12, 24]. Moreover, the PCR method for the specific detection of MDPV requires high precision primer design. Loop-mediated isothermal amplification (LAMP) [25] and an aptamer by label-free aptasensor [26] against MDPV for a highly sensitive, rapid visual detection was designed that targeted the VP3 gene of MDPVs. The VP3 protein is the most variable and abundant protein of MDPV. VP3 can induce neutralizing antibodies in GPV- or MDPV-infected waterfowl and confers protective cross-immunity in waterfowl parvoviruses [13, 27,28,29,30]. Moreover, VP3 has been associated with many genetic variations because of virus evolution selection pressure [6, 7, 31,32,33]. In addition, due to lack of information on the complete genomic sequence data of GPVs, especially MDPVs, false results can be obtained if primers are designed from unsuitable regions.
The real-time PCR technology has advantages with its remarkable sensitivity, specificity, reproducibility, visualization results, time-saving benefits, high-throughput analysis and less-contamination potential compared to other diagnostic methods, that have been widely used for viral pathogenesis research and epidemiology surveillance [16,17,18,19,20,21]. Woźniakowski et al. developed a TaqMan-based real-time PCR method for GPVs and MDPVs in which the designed primers and TaqMan probe for real-time PCR were complementary to GPV and MDPV inverted terminal repeats region (ITR) [34]. The calculation of results is often confounded due to the two ITR repeat regions at the 5′- and 3′-terminus in both the GPV and MDPV genomes. Moreover, mutation and deletion have been found in ITR of both GPVs and MDPVs, which may cause high risk of failure for the specific detection of MDPV infection [35,36,37]. In this study, we compared a total of 52 NS gene sequences (including 15 MDPVs and 37 GPVs) retrieved from the GenBank. We found that within the MDPV cluster, the samples shared higher nucleotide identity (more or equal than 98.0%) than within GPV cluster (more or equal than 93.3%). In addition, between GPV cluster and MDPV cluster, the NS gene homology ranged from 80.8 to 83.4%. These data indicate that false results may be obtained if these primers are designed at the NS gene-specific regions. These NS genetic comparison data allowed us to identify the MDPV-specific suitable regions, which can be used to establish a TaqMan-based real-time PCR assay for the detection of MDPV with more precision.
In this study, we clearly developed and evaluated the applicability of TaqMan-based real-time PCR for the detection and quantification of MDPV. The data of the qPCR assay was performed over a wide dynamic range with no fluorescence signal can be observed from other duck-derived pathogens, and low intra- and inter-assay variation (less than 2.21%). Field samples from cloacal swabs detection and those used in previous studies of MDPV infection provided the evidence that MDPV can be horizontally transmitted. Furthermore, the positive fluorescence signals observed in embryos and newly hatched Muscovy ducklings support the view that MDPV could be possible vertically transmitted as well as N-GPV [23].
Conclusions
A specific and reliable TaqMan-based real-time PCR for the detection and quantification of MDPV was developed, because of significantly conserved region of NS genes between MDPVs and GPVs were selected for primers and probe design. The detection of the MDPV-positive in Muscovy duck embryos and newly hatched Muscovy ducklings, provide evidence of possible vertical transmission of MDPV from breeding Muscovy ducks to Muscovy ducklings.
Abbreviations
- AIV:
-
avian influenza virus;
- ATmV:
-
avian Tembusu virus
- BLAST:
-
Basic Local Alignment Search Tool
- cPCR:
-
conventional PCR
- Ct:
-
threshold cycle
- CV:
-
Coefficient of variation
- DAdV-A:
-
Duck adenovirus A
- DEV:
-
Duck enteritis virus
- DHAV:
-
Duck hepatitis virus
- GPV:
-
Goose parvovirus
- ICTV:
-
International Committee on Taxonomy of Viruses
- ITR:
-
Inverted terminal repeats
- LAMP:
-
Loop-mediated isothermal amplification
- MDPV:
-
Muscovy duck parvovirus
- MDRV:
-
Muscovy duck reovirus
- N-GPV:
-
Novel goose parvovirus
- NS:
-
Nonstructural
- ORF:
-
Open reading frame
- P.M. :
-
Pasteurellamultocida
- qPCR:
-
real-time quantitative PCR
- R.A. :
-
Rimerella anatipstifer
- R2 :
-
correlation
- S.S. :
-
Salmonella spp
References
Zadori Z, Erdei J, Nagy J, Kisary J. Characteristics of the genome of goose parvovirus. Avian Pathol. 1994;23:359–64.
Zádori Z, Stefancsik R, Rauch T, Kisary J. Analysis of the complete nucleotide sequences of goose and muscovy duck parvoviruses indicates common ancestral origin with adeno-associated virus 2. Virology. 1995;212:562–73.
Le Gall-Reculé G, Jestin V. Biochemical and genomic characterization of muscovy duck parvovirus. Arch Virol. 1994;139:121–31.
Chen H, Dou Y, Tang Y, Zhang Z, Zheng X, Niu X, Yang J, Yu X, Diao Y. Isolation and genomic characterization of a duck-origin GPV-related parvovirus from Cherry Valley ducklings in China. PLoS One. 2015;10:e0140284.
Liu H, Wang H, Tian X, Zhang S, Zhou X, Qi K, Pan L. Complete genome sequence of goose parvovirus Y strain isolated from Muscovy ducks in China. Virus Genes. 2014;48:199–202.
Zhu Y, Zhou Z, Huang Y, Yu R, Dong S, Li Z, Zhang Y. Identification of a recombinant Muscovy duck parvovirus (MDPV) in shanghai, China. Vet Microbiol. 2014;174:560–4.
Wang J, Ling J, Wang Z, Huang Y, Zhu J, Zhu G. Molecular characterization of a novel Muscovy duck parvovirus isolate: evidence of recombination between classical MDPV and goose parvovirus strains. BMC Vet Res. 2017;13:327.
Wan C, Chen H, Fu Q, Fu G, Cheng L, Shi S, Huang Y, Hu K. Genomic characterization of goose parvovirus and Muscovy duck parvovirus co-infection in Fujian. China Kafkas Univ Vet Fak Derg. 2015;21:923–8.
Shao H, Lv Y, Ye J, Qian K, Jin W, Qin A. Isolation of a goose parvovirus from swan and its molecular characteristics. Acta Virol. 2014;58:194–8.
Wan C, Huang Y, Shi S, Fu G, Cheng L, Chen H, Fu Q, Chen C, Hu K. Complete genome sequence of goose parvovirus isolated from Anser cygnoides in China. Kafkas Univ Vet Fak Derg. 2016;22:155–8.
Lin S, Yu X, Chen B, Chen B. The diagnosis of a new muscovy duck virus infection. Chin J Prevent Vet Med. 1991;2:27–8. (in Chinese)
Wan C, Chen H, Fu Q, Shi S, Fu G, Cheng L, Chen C, Huang Y, Hu K. Development of a restriction length polymorphism combined with direct PCR technique to differentiate goose and Muscovy duck parvoviruses. J Vet Med Sci. 2016;78:855–8.
Zhang Y, Li Y, Liu M, Zhang D, Guo D, Liu C, Zhi H, Wang X, Li G, Li N, Liu S, Xiang W, Tong G. Development and evaluation of a VP3-ELISA for the detection of goose and Muscovy duck parvovirus antibodies. J Virol Methods. 2010;163:405–9.
Li M, Yu TF. Immunologic cross-reactivity between Muscovy duck parvovirus and goose parvovirus on the basis of epitope prediction. Braz J Microbiol. 2013;44:519–21.
Yu TF, Li M, Yan B, Shao SL, Fan XD, Wang J, Wang DN. Identification of antigenic domains in the non-structural protein of Muscovy duck parvovirus. Arch Virol. 2016;161:2269–72.
Niesters HG. Quantitation of viral load using real-time amplification techniques. Methods. 2001;25:419–29.
Mackay IM, Arden KE, Nitsche A. Real-time PCR in virology. Nucleic Acids Res. 2002;30:1292–305.
Niu X, Chen H, Yang J, Yu X, Ti J, Wang A, Diao Y. Development of a TaqMan-based real-time PCR assay for the detection of novel GPV. J Virol Methods. 2016;237:32–7.
Wang J, Wang J, Cui Y, Nan H, Yuan W. Development of a taqman-based real-time PCR assay for the rapid and specific detection of novel duck- origin goose parvovirus. Mol Cell Probes. 2017;34:56–8.
Wan C, Cheng L, Fu G, Chen C, Liu R, Shi S, Chen H, Fu Q, Huang Y. Rapid detection of goose hemorrhagic polyomavirus using TaqMan quantitative real-time PCR. Mol Cell Probes. 2018;39:61–4.
Wan C, Chen C, Cheng L, Guanghua F, Shi S, Liu R, Chen H, Fu Q, Huang Y. Development of a TaqMan-based real-time PCR for detecting duck adenovirus 3. J Virol Methods. 2018; https://doi.org/10.1016/j.jviromet.2018.08.011.
Yun JJ, Heisler LE, Hwang II, Wilkins O, Lau SK, Hyrcza M, Jayabalasingham B, Jin J, McLaurin J, Tsao MS, Der SD. Genomic DNA functions as a universal external standard in quantitative real-time PCR. Nucleic Acids Res. 2006;34:e85.
Chen H, Tang Y, Dou Y, Zheng X, Diao Y. Evidence for vertical transmission of novel duck-origin goose parvovirus-related parvovirus. Transbound Emerg Dis. 2016;63:243–7.
Sirivan P, Obayashi M, Nakamura M, Tantaswasdi U, Takehara K. Detection of goose and Muscovy duck parvoviruses using polymerase chain reaction-restriction enzyme fragment length polymorphism analysis. Avian Dis. 1998;42:133–9.
Ji J, Xie QM, Chen CY, Bai SW, Zou LS, Zuo KJ, Cao YC, Xue CY, Ma JY, Bi YZ. Molecular detection of Muscovy duck parvovirus by loop-mediated isothermal amplification assay. Poult Sci. 2010;89:477–83.
Lu T, Ma Q, Yan W, Wang Y, Zhang Y, Zhao L, Chen H. Selection of an aptamer against Muscovy duck parvovirus for highly sensitive rapid visual detection by label-free aptasensor. Talanta. 2018;176:214–20.
Le Gall-Reculé G, Jestin V, Chagnaud P, Blanchard P, Jestin A. Expression of muscovy duck parvovirus capsid proteins (VP2 and VP3) in a baculovirus expression system and demonstration of immunity induced by the recombinant proteins. J Gen Virol. 1996;77:2159–63.
Yin X, Zhang S, Gao Y, Li J, Tan S, Liu H, Wu X, Chen Y, Liu M, Zhang Y. Characterization of monoclonal antibodies against waterfowl parvoviruses VP3 protein. Virol J. 2012;9:288.
Tarasiuk K, Woźniakowski G, Holec-Gąsior L. Expression of goose parvovirus whole VP3 protein and its epitopes in Escherichia coli cells. Pol J Vet Sci. 2015;18:879–80.
Li C, Liu H, Li J, Liu D, Meng R, Zhang Q, Shaozhou W, Bai X, Zhang T, Liu M, Zhang Y. A conserved epitope mapped with a monoclonal antibody against the VP3 protein of goose parvovirus by using peptide screening and phage display approaches. PLoS One. 2016;11:e0147361.
Chu CY, Pan MJ, Cheng JT. Genetic variation of the nucleocapsid genes of waterfowl parvovirus. J Vet Med Sci. 2001;63:1165–70.
Wang S, Cheng X, Chen S, Zhu X, Chen S, Lin F, Li Z. Genetic characterization of a potentially novel goose parvovirus circulating in Muscovy duck flocks in Fujian Province, China. J Vet Med Sci. 2013;75:1127–30.
Wang S, Cheng X, Chen S, Lin F, Chen S, Zhu X, Wang J. Evidence for natural recombination in the capsid gene VP2 of Taiwanese goose parvovirus. Arch Virol. 2015;160:2111–5.
Woźniakowski G, Samorek-Salamonowicz E, Kozdruń W. Quantitative analysis of waterfowl parvoviruses in geese and Muscovy ducks by real-time polymerase chain reaction: correlation between age, clinical symptoms and DNA copy number of waterfowl parvoviruses. BMC Vet Res. 2012;8:29.
Shien JH, Wang YS, Chen CH, Shieh HK, Hu CC, Chang PC. Identification of sequence changes in live attenuated goose parvovirus vaccine strains developed in Asia and Europe. Avian Pathol. 2008;37:499–505.
Wang J, Duan J, Zhu L, Jiang Z, Zhu G. Sequencing and generation of an infectious clone of the pathogenic goose parvovirus strain LH. Arch Virol. 2015;160:711–8.
Wang J, Huang Y, Zhou M, Zhu G. Analysis of the genome sequence of the pathogenic Muscovy duck parvovirus strain YY reveals a 14-nucleotide-pair deletion in the inverted terminal repeats. Arch Virol. 2016;161:2589–94.
Funding
This work was funded by the Natural Science Foundation of China (31602068), China Agriculture Research System (CARS-42), Fujian Academy of Agriculture Science Innovative Research Team Project (STIT2017-3-10) and Young Talent Program Project (YC2015–12), and the Fujian Public Welfare Project (2018R1023–5). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article. All datasets are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
CW assembled the sequence data, designed primers and performed the experiments, analyzed the data and drafted the manuscript. CC, LC, HC, QF, SS, GF, and RL collected samples and helped in laboratory analysis. YH contributed to experimental design and supervised the study. All authors read, commented on and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The animal protocol used in this study was approved by the Research Ethics Committee of the College of Institute of Animal Husbandry and Veterinary Medicine, Fujian Academy of Agriculture Sciences (Permit Number FAAS-AHVM2017–18). All ducks were handled in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wan, C., Chen, C., Cheng, L. et al. Specific detection of Muscovy duck parvovirus infection by TaqMan-based real-time PCR assay. BMC Vet Res 14, 267 (2018). https://doi.org/10.1186/s12917-018-1600-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-018-1600-3