
Abstract
Treatment of multidrug-resistant tuberculosis (MDR-TB) is extremely challenging due to the virulence of the etiologic strains of Mycobacterium tuberculosis (M. tb), the aberrant host immune responses and the diminishing treatment options with TB drugs. New treatment regimens incorporating therapeutics targeting both M. tb and host factors are urgently needed to improve the clinical management outcomes of MDR-TB. Host-directed therapies (HDT) could avert destructive tuberculous lung pathology, facilitate eradication of M. tb, improve survival and prevent long-term functional disability. In this review we (1) discuss the use of HDT for cancer and other infections, drawing parallels and the precedent they set for MDR-TB treatment, (2) highlight preclinical studies of pharmacological agents commonly used in clinical practice which have HDT potential, and (3) outline developments in cellular therapy to promote clinically beneficial immunomodulation to improve treatment outcomes in patients with pulmonary MDR-TB. The use of HDTs as adjuncts to MDR-TB therapy requires urgent evaluation.
Similar content being viewed by others

Background
The World Health Organization (WHO) presently ranks tuberculosis (TB) as the leading cause of death due to infectious disease worldwide, with an estimated 1.5 million deaths from TB in 2014 [1]. Multidrug-resistant TB (MDR-TB) accounted for more than 3 % of newly diagnosed TB cases, and over 10 % (190,000) of all TB-related deaths [1]. Further, 12 % of individuals diagnosed with TB were also co-infected with HIV, comprising more than 20 % (390,000) of deaths due to TB in 2014 [1]. Current WHO guidelines for MDR-TB treatment recommend the use of combination TB drug therapies that target Mycobacterium tuberculosis (M. tb). These drugs, several of which have serious side effects, have to be taken for a period of 12–18 months, with the long duration resulting in poor patient compliance. Furthermore, current MDR-TB treatment regimens cure only 50 % of MDR-TB cases and impose a huge financial toll on national health services [1, 2]. Other important limitations of current MDR-TB treatment include drug interactions and insufficient drug concentrations in patients’ lungs [3] as well as aberrant or excess host inflammatory responses to M. tb, resulting in lung tissue destruction, compromising the body’s ability to eradicate the M. tb bacilli, and leading to permanent lung tissue damage [4–6].
Pulmonary pathology observed in patients with all forms of TB can be attributed to intrinsic exaggerated inflammatory responses to M. tb [4]. Since HIV co-infection is the major comorbidity associated with poor clinical outcome in patients with active TB, early restoration of immune cells in HIV-TB patients following antiretroviral therapy (ART) can result in the development of immune reconstitution inflammatory syndrome (TB-IRIS) [5]. In TB-IRIS, existing M. tb-specific T cells proliferate uncontrollably, consequently perpetrating local and systemic overt immune reactions and potentially leading to death [5]. An exaggerated release of pro-inflammatory cytokines, such as tumour necrosis factor alpha (TNF-α), interleukin 6 (IL-6), IL-8, IL-18, and interferons alpha and beta (IFN-α/β), invariably perpetrate tissue damage in active TB as well as TB-IRIS [6, 7]. Although TB-IRIS is more closely associated with drug-susceptible TB patients starting on ART, it has been reported in South African patients with MDR-TB and HIV co-infection [8]. An exigent need therefore exists to develop new strategies to treat and prevent harmful inflammatory responses in MDR/extensively drug resistant-TB patients to avoid long-term, if not permanent, lung damage [9–12].
Considering these limitations and intricate clinical scenarios in TB, host-directed therapies (HDTs) have emerged as a new and promising intervention avenue against all forms (drug-sensitive and drug-resistant) of the disease [13]. HDTs generally target clinically relevant biological pathways in the host to modulate and rectify pathological immune responses. In TB, HDTs may neutralise excessive inflammation in organs and decrease M. tb proliferation while facilitating tissue repair [7, 10].
Therapeutic targeting of immunological and cellular processes in MDR-TB
Over the past several years, extensive preclinical and clinical studies have revealed a myriad of immunological pathways and biomarkers that either influence the outcome of M. tb infection or indicate the state of disease, respectively [6, 14–18]. These pathways include cytokine-mediated signalling, intracellular antimicrobial processes, and establishment of long-term immunological memory as well as physiological homeostasis [13]. The use of immunomodulatory agents as HDTs for TB treatment has therefore garnered great interest as a means of improving clinical outcome of therapy, based on previous experience with other infectious diseases and cancer. Many of these agents are already licensed for non-TB indications, thus bearing US Food and Drug Administration approval, while others are in advanced clinical trials [18, 19]. A summary of the various HDT agents with therapeutic potential in pulmonary MDR-TB is presented in Table 1 and Fig. 1.

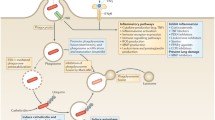
Host-directed therapies aimed at modulating immune responses in the tuberculous lung. Overt immune responses characterise the pathological outcome in tuberculosis (TB). Neutralisation of pro-inflammatory cytokines such as IL-6, TNF-α, VEGF and IFN-αβ, as well as anti-inflammatory IL-4 during severe pulmonary disease may help reduce ongoing parenchymal damage in the lung. Alternatively, suboptimal activation of anti-TB immune responses due to regulatory T cell activity can be reversed by the use of the anti-cancer drug cyclophosphamide. Drugs with anti-TB potential, such as metformin, imatinib, ibuprofen, zileuton, valproic acid, and vorinostat as well as nutraceuticals such as vitamin D3 not only abate bacterial burden via host-dependent mechanisms, but may also fine-tune the immune response to Mycobacterium tuberculosis (M. tb). These drugs increase phagocytosis of extracellular bacteria, improved emergency myeloid response and increased autophagic and apoptotic killing of bacteria, subsequently editing the T cell response in favour of the host. Immune checkpoint inhibition with blockade of the PD-1/PD-L1, CTLA-4, LAG3 and TIM3 pathways may improve the quality of the cellular immune response to M. tb epitopes, as seen in cancer. A more complete list of currently pursued host-directed therapies for TB can be found in Table 1. Abbreviations: VPA, valproic acid; PBA, phenylbutyrate; PD-1, programmed cell death 1; PD-L1, PD-1 ligand 1; CTLA-4, cytotoxic T lymphocyte-associated antigen 4; LAG3, lymphocyte-activation gene 3; TIM3, T cell immunoglobulin and mucin domain 3
Immunomodulatory small-molecule drugs
Several non-antibiotic drugs with immunomodulatory properties have already shown clinical efficacy in ameliorating infection-induced pathology. For example, interventional use of glucocorticoid receptor agonists, i.e., prednisone and dexamethasone, has mortality-reducing effects in community-acquired pneumonia, concomitant with lowering of pro-inflammatory cytokine release [20, 21]. However, the clinical outcome of prednisone therapy in pulmonary TB has been inconclusive; further studies are required to ascertain a clear beneficial effect in patients [22].
Eicosanoid regulation dependent on arachidonic acid (ARA) metabolism has been associated with modulation of clinically relevant immune responses and pathology in patients with active TB [23]. Two major arms of enzymatic reactions carry out ARA catabolism: the first involving cyclooxygenases 1 and 2 (COX1 and 2) and the other, lipoxygenases (LO). Breakdown of ARA by COX enzymes progresses to prostaglandin synthesis, while LO metabolise ARA to produce leukotrienes, lipid mediators responsible for fever induction during active infection [24]. COX2 is particularly associated with regulation of pro-inflammatory cytokine production and inflammation via prostaglandin E2 (PGE2) production [25]. Leukotrienes are also involved in initiation of inflammation, angiogenesis and progression of cancer [25].
It has been shown in TB that balance between concentrations of lipoxin A4 (LXA4) and leukotrienne B4 controls subsequent TNF-α release by activated immune cells in the lung [23]. Overproduction of LXA4 may instead cause immunosuppression, highlighting that maintenance of this axis in TB is pivotal in determining patient survival or death. The use of aspirin (acetylsalicylic acid), a non-steroidal anti-inflammatory drug (NSAID) inhibits COX2 activity, thus enhancing LXA4 synthesis, which in turn would decrease TNF-α levels and subsequently neutralise pulmonary inflammation in TB patients [23, 26]. The same may be achieved with ibuprofen (another NSAID), which has been shown to improve survival of experimental mice with severe lung TB by dampening tissue pathology while reducing M. tb load [27]. Ibuprofen also has direct anti-M. tb activity [28].
Zileuton is an anti-inflammatory drug currently licensed for the treatment of asthma that exerts its effects by inhibiting 5-LO activity [29]. As opposed to NSAIDs, zileuton blocks LO-mediated leukotriene production, facilitating COX2 activation and PGE2 synthesis. In a mouse model of TB, zileuton was shown to dramatically abate IFN-α/β levels in the lung to improve lung pathology, in addition to reducing M. tb load [30]. Combination therapy comprising zileuton and PGE2 augmented interleukin 1 beta (IL-1β)-mediated control of M. tb growth. Furthermore, patients with latent TB infection who successfully contained infection and did not develop active pulmonary TB appear to maintain balanced serum levels of PGE2 and LXA4 [30].
In addition to their cholesterol-lowering properties, statins also possess anti-inflammatory properties, which enhance their efficacy in the clinical management of atherosclerosis [31]. Statins can reduce IFN-γ-mediated MHC-II upregulation, and thus CD4 T cell activation – having clinical benefits in cardiac transplantation [32]. Statins may also downregulate TNF-α and IL-1β release via activation of the transcription factor Kruppel-like factor 2 (KLF2), as well as reduce IL-6, IL-8 and CCL5 secretion by myeloid cells and lymphocytes [31]. Prior use of statins has been shown to ameliorate inflammation-induced organ damage, thus reducing mortality among patients with sepsis and community-acquired pneumonia [33–36]. The therapeutic potential of statins has been evaluated in TB. Peripheral blood mononuclear cells derived from patients with familial hypercholesterolemia undergoing atorvastatin therapy were found to be more resistant to M. tb infection and replication [37]. It was established in a mouse model of TB that the cholesterol-lowering properties of atorvastatin potentiated this effect, where simvastatin treatment was concomitant with reduced pulmonary, hepatic and splenic M. tb burden and tissue pathology [37]. It was also shown that simvastatin potentiates autophagic cell death of M. tb-infected macrophages, thus having a dual approach in controlling TB disease. In a separate study, co-administration of simvastatin to an anti-TB chemotherapy regimen combining rifampicin, isoniazid and pyrazinamide led to improved bactericidal activity of the drugs against intracellular M. tb as well as in a mouse model of TB [38]. Clinically, patients with type 2 diabetes mellitus (T2DM) on statin therapy (as well as anti-diabetic therapy) seem to be at lower risk of developing active TB [39].
Metformin (MET) is an essential drug used in the treatment of T2DM, which acts by affecting the mitochondrial respiratory chain [40]. In a recent preclinical study, MET was evaluated as an adjunct therapeutic for drug-susceptible (DS)-TB and MDR-TB [41]. MET treatment induced superoxide anion production in macrophages infected with DS or MDR M. tb in an-adenosine monophosphate-activated protein kinase (AMPK)-dependent manner [41]. Furthermore, MET therapy potentiated the activity of the first-line anti-TB drugs isoniazid and ethionamide in a mouse model of TB, commensurate with improved anti-TB CD8 T cell responses, reduced concentrations of inflammatory cytokines and lung pathology [41]. A retrospective evaluation of patients with T2DM receiving MET therapy showed that they are less prone to develop cavitary pulmonary TB. In addition, T cell non-reactivity to ESAT-6 in a T-SPOT IFN-γ assay also suggested less incidence of latent TB infection among these individuals [41]. The ability of MET to promote maintenance of memory CD8 T cells via AMPK-driven fatty acid oxidation, as seen in mouse models of solid tumours [42, 43], may be an additional avenue for its potent immunomodulatory properties in TB.
Imatinib mesylate, a highly specific and successful drug that inhibits the Bcr-Abl tyrosine kinase in chronic myelogenous leukaemia has also been evaluated in TB [44]; treatment of M. marinum-infected mice with imatinib resulted in enhanced myelopoiesis in the bone marrow. Furthermore, an early influx of neutrophils into the lungs of infected mice helped reduce the bacterial burden. Imatinib in combination with rifampicin greatly reduced liver pathology in mice infected with M. marinum, while reducing lung bacterial load also in M. tb-infected animals [45].
The DNA alkylating agent cyclophosphamide (CP) is a potent anti-proliferative cancer drug and pre-conditioning agent metabolised by cytochrome P450 to produce reactive metabolites that crosslink with guanine residues [46, 47]. Cell types expressing low amounts of aldehyde dehydrogenase are most susceptible to CP treatment since its enzymatic activity neutralises drug-derived cytotoxicity [48]. Regulatory T cells (Tregs) express high cellular levels of ATP-binding cassette B1, which binds CP, thus making these cells especially vulnerable to the drug [49]. Conversely, aldehyde dehydrogenase overexpression can promote resistance to CP toxicity [50]. Prior CP administration to patients participating in a clinical trial of a renal cell cancer vaccine candidate (IMA901) led to Treg decline in peripheral blood of the participants, afforded better CD8 T cells priming and improved overall survival [51]. CP treatment may remove Treg cells as well as pathological lymphocytosis in patients with MDR-TB [52–54].
Histone deacetylase inhibitors (HDIs) comprise several clinically approved pharmacological agents which block the activity of the chromosome-modifying enzymes histone deacetylases to promote gene transcription [55]. Histone acetylation mediated by HDIs has been clinically beneficial in cancer therapy [56], vorinostat (suberoylanilide hydroxamic acid) was licensed in 2006 for the treatment of cutaneous T cell lymphoma [57], and valproic acid (VPA, currently in use for epilepsy and bipolar disorder) is in clinical trials for solid tumours [58], while many others are in late-stage clinical trials for a plethora of cancers [59]. VPA and vorinostat can induce apoptosis and autophagy in treated cells via oxidative burst and inhibition of the mammalian target of rapamycin (mTOR), respectively [60–62]. The effectiveness of HDIs in infectious disease has been evaluated against HIV infection and West Nile virus infection. VPA or vorinostat treatment of CD4 T cells from individuals latently infected with HIV-1 can reactivate viral progeny, making virus-infected cells highly susceptible to ART as well as CD8 T cell-mediated immune attack [63–66]. These observations have led to investigating the clinical efficacy of HDIs in HIV-infected individuals [67, 68]. In a mouse model of West Nile virus infection, co-treatment of vorinostat with an experimental antiviral drug candidate ameliorated brain inflammation and neuronal damage in addition to reduction in viral load [69]. Evaluation of phenylbutyrate (PBA), another HDI, against intracellular M. tb has been shown to potentiate pro-inflammatory signalling (upregulation of CASP1, IL12, and VDR genes, among others), antimicrobial peptide production, reduced M. tb survival, thus implying enhanced anti-TB T cell activity [70]. Clinical evaluation of PBA and vitamin D3 treatment in patients with pulmonary TB resulted in quicker sputum smear conversion and increased production of cathelicidin by peripheral blood monocyte-derived macrophages [71].
Blockade of cytokine signalling
As previously mentioned, the uncontrolled release of pro-inflammatory cytokines, such as TNF-α, IL-6 and IFN-α/β, drives destructive immunopathology and severe lung disease in TB. The use of anti-TNF-α therapy (adalimumab) in a patient with severe pulmonary TB proved to be life-saving [72], and set the stage for anti-cytokine therapy in clinical management of TB. IL-6, a major player in exacerbating disease severity in patients with TB, is another potential candidate for clinical blockade [6, 73], further confirmed by reduction of IL-6 serum levels in TB patients post-treatment [74]. Preclinical evaluation of IL-6 receptor (IL-6R) blockade in a mouse model of TB improved specific anti-TB T cell responses in conjunction with less severe pathology and reduced M. tb burden in the lungs [75]. Monoclonal antibodies that inhibit the IL-6/IL-6R pathway are currently endorsed for treatment of immune-mediated diseases such as idiopathic juvenile arthritis and Castleman’s disease in humans (anti-IL-6, siltuximab; anti-IL-6R, tocilizumab) [76, 77], and are currently under investigation for cancer treatment [78]. Blockade of the IL-6/IL-6R pathway may, therefore, be considered for clinical management of severe pulmonary MDR-TB. Preclinical evaluation of bevacizumab (anti-vascular endothelial growth factor (VEGF) monoclonal antibody) in a rabbit model of TB showed that inhibition of VEGF-A was commensurate with modified angiogenesis in the granuloma, which improved penetration of a small molecule dye (representative of an anti-TB drug) as well as oxygenation [79]. Oehlers et al. [80] showed that VEGF promotes granuloma formation in M. marinum-infected zebrafish, while pharmacological inhibition of VEGF activity with pazopanib reduced neovascularisation as well as the mycobacterial burden in the animals. IL-4 is another cytokine associated with poor prognosis in clinical TB that quantitatively declines in response to anti-TB treatment [81–84]. A clinical trial investigating the adjunctive potential of IL-4 blockade (anti-human IL-4 monoclonal antibody, pascolizumab) in patients with DS pulmonary TB undergoing anti-TB treatment is currently underway (NCT01638520).
Vitamin D3
Vitamin D3 (VD3) is important for host resistance to TB; pulmonary TB patients with VD3 deficiency are not able to mount adequate control of primary M. tb infection in the lung [85]. Further, VD3 has been implicated in immunological control of TB in humans involving orchestration of IFN-γ, IL-32 and IL-15 signalling [86, 87]. Recent evaluation of the therapeutic potential of VD3 in combination with PBA has shown improved anti-mycobacterial effects and faster conversion to AFB-negative sputum by the participating TB patients [71]. In vitro evaluation of M. tb-infected macrophages treated with VD3 resulted in upregulation of a collage of anti-inflammatory (IL-10, ARG1) and pro-inflammatory (IL1B, TNF) genes over a 72-hour exposure period [70] – this profile was relatively unchanged with PBA co-treatment. However, other clinical studies have not found a correlation between VD3 therapy and improved treatment outcome in TB patients [88, 89]. The therapeutic potential of VD3 in MDR-TB patients therefore warrants further clinical evaluation in larger cohorts of patients in endemic countries.
Immune checkpoint inhibition
One of the most remarkable immunomodulatory therapies of recent times is currently licensed for melanoma treatment – antibody-based inhibition of the immune checkpoints programmed cell death 1 (PD-1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4). This strategy activates and mobilises tumour-infiltrating T lymphocytes as well as those in peripheral blood of patients with metastatic melanoma [90]. Although immunologically purposed to regulate T cell responses for avoiding immune hyper reactivity, PD-1 and CTLA-4 expression on tumour-specific T cells abrogates anti-tumour effector functions and renders the T cells irresponsive [91]. Tregs have abundant expression of PD-1 on their surface; interaction of this molecule with its ligands (PD-L1/2) on target cells leads to expansion and dampening of antigen-specific effector T cell responses [92]. Importantly, a myriad of cytokines, including IFN-γ, TNF-α, type 1 interferons, GM-CSF, IL-2, IL-7 and IL-15, are actively involved in enhancing the expression of PD-1 on T cells and its ligands on antigen-presenting cells and tumour cells at sites of disease [93]. CTLA-4 binds to CD80 and/or CD86 to control the activation state of effector T cells in the aftermath of a successful immune response, and initiates tolerance by abrogating IL-2 production and downstream activation of signal transducer and activator of transcription 5 (STAT5) [94].
Results from clinical trials assessing monoclonal antibodies targeting PD-1 (pembrolizumab/nivolumab) and CTLA-4 (ipilimumab) in cancers other than melanoma, i.e., lung cancer and ovarian cancer, have been very promising [95, 96]. Anti-PD-1 therapy (nivolumab) has been fruitful also in treating patients with chronic hepatitis C virus infection, resulting in a dramatic reduction in viral load [97]. There are several ongoing clinical studies evaluating the efficacy of anti-PD-L1 therapy (avelumab, MEDI4736) in cancer (e.g., but not limited to, NCT02572843, NCT02584829, NCT02588131, NCT02088112). Several clinical studies have evaluated the role of the PD-1/PD-L1 pathway in the pathogenesis of TB. PD-L1, the main ligand for PD-1, is markedly expressed on human macrophages upon infection with virulent M. tb [98]. In vitro blockade of PD-L1 with monoclonal antibodies induced IFN-γ-triggered killing of the infected cells by autologous peripheral blood T cells from patients with pulmonary TB [98, 99]. Clinically, PD-1 expression on T cells declines over a course of standard anti-TB therapy – concordant with improved in vitro responsiveness of Th1 cells to antigenic stimulation [99, 100]. In addition, expression of PD-1 on the surface of neutrophils, Tregs and natural killer T cells indicates that destructive inflammation in TB patients may further impair the M. tb-specific immune response [53, 54, 101, 102]. Similar observations have been reported for CTLA-4 expression on Tregs and effector T cells isolated from peripheral blood of patients with active TB [53, 103, 104].
Lymphocyte-activation gene 3 (LAG3) is another immune checkpoint generally expressed by Tregs to modulate CD4+ T cell responses, and binds to MHC-II molecules with high affinity to induce T cell exhaustion several days after activation [105]. LAG3 was recently shown to be highly expressed in the lungs of macaques with active pulmonary TB and not in latent TB infection, suggesting a link between low LAG3 expression and successful containment of M. tb infection [106]. Combined blockade of PD-1 and LAG3 is able to rescue specific CD4+ and CD8+ T cell activity directed against Plasmodium falciparum in an experimental mouse model of malaria [95, 107]. Several clinical trials are currently registered for testing the efficacy of anti-LAG3 monoclonal antibodies (BMS-986016, GSK2831781, LAG525) in various cancers (NCT02061761, NCT01968109, NCT02195349, NCT02460224).
The fourth clinically relevant immune checkpoint molecule is T cell immunoglobulin and mucin domain 3 (TIM3). Immunologically, TIM3 binds to galectin 9 (Gal9) on the surface of antigen-presenting cells (as well as tumour cells) to induce apoptosis of activated T cells [108]. Inhibition of TIM3 (MBG453) in cancer therapy affords improvement of anti-tumour T cell responses, marked by IFN-γ secretion and CD8 T cell effector functions [108]. Pertinent to TB, in vitro TIM3 blockade in co-culture experiments with M. tb-infected macrophages from TB patients with or without HIV co-infection fostered bacterial killing and enhanced IL-1β secretion by infected cells as well as IFN-γ release by T cells [109, 110]. T cell-mediated reduction of bacterial burden in infected macrophages from HIV-TB patients was further improved in combination with anti-PD-1 blockade [110]. Along with PD-1 and CTLA-4, treatments targeting LAG3 and TIM3 may improve targeted immune responses in patients with MDR-TB for better clinical outcomes.
Cellular therapy
In addition to chemical compounds, monoclonal antibodies and nutraceuticals, cell-based therapies have also been very promising in cancer and infectious disease [111, 112]. Adoptive transfer of tumour-infiltrating T lymphocytes has saved many lives in the last 20 years, with objective response rates reaching 56 % in patients with metastatic melanoma [113]. Transfer of donor-derived cytomegalovirus- and Epstein–Barr virus-specific CD8 T cells following stem cell transplantation is actively pursued in the clinic [111]. Pertaining to TB, re-infusion of bone marrow-derived mesenchymal stromal cells isolated from patients with MDR/extensively drug resistant-TB was shown to be safe [114], and a phase 2 trial is underway in Durban, South Africa, to evaluate their efficacy in improving treatment outcomes and effects on immune responses. Mesenchymal stromal cell treatment also re-programmed anti-TB T cell responses, with focus on specific M. tb antigens. Infusion of M. tb-specific T cells has yet to be clinically evaluated in TB patients, although in vitro evidence for feasibility, comprising subsets of CD8 T cells, NK T cells and TCR γδ T cells, has been established [115–119].
Conclusion
The poor treatment outcomes of MDR-TB treatment using current WHO recommended TB drug treatment regimens now warrants newer approaches to therapy. The use of immunomodulatory agents as adjunct HDT for improving management outcomes warrants urgent evaluation in well designed, controlled randomized clinical trials. These studies will also provide opportunities to study the M. tb-specific immune responses in the lung and peripheral blood (B cells, T cells, macrophages, antibodies) as markers of inflammation, disease activity, cure or relapse. Well-designed, multicentre randomised clinical trials involving aptly defined patient cohorts are now required.
References
WHO. Global Tuberculosis Report 2015. Geneva: World Health Organization; 2015. p. 204.
Schnippel K, Rosen S, Shearer K, Martinson N, Long L, Sanne I, Variava E. Costs of inpatient treatment for multi-drug-resistant tuberculosis in South Africa. Trop Med Int Health. 2013;18(1):109–16.
Dheda K, Gumbo T, Gandhi NR, Murray M, Theron G, Udwadia Z, Migliori GB, Warren R. Global control of tuberculosis: from extensively drug-resistant to untreatable tuberculosis. Lancet Respir Med. 2014;2(4):321–38.
Zumla A, Rao M, Parida SK, Keshavjee S, Cassell G, Wallis R, Axelsson-Robertsson R, Doherty M, Andersson J, Maeurer M. Inflammation and tuberculosis: host-directed therapies. J Intern Med. 2015;277(4):373–87.
Lai RP, Meintjes G, Wilkinson RJ. HIV-1 tuberculosis-associated immune reconstitution inflammatory syndrome. Semin Immunopathol. 2015. doi:10.1007/s00281-015-0532-2.
Ravimohan S, Tamuhla N, Steenhoff AP, Letlhogile R, Nfanyana K, Bellamy SL, MacGregor RR, Gross R, Weissman D, Bisson GP. Immunological profiling of tuberculosis-associated immune reconstitution inflammatory syndrome and non-immune reconstitution inflammatory syndrome death in HIV-infected adults with pulmonary tuberculosis starting antiretroviral therapy: a prospective observational cohort study. Lancet Infect Dis. 2015. doi:10.1016/S1473-3099(15)70008-3.
Zumla A, Rao M, Parida SK, Keshavjee S, Cassell G, Wallis R, Axelsson-Robertsson R, Doherty M, Andersson J, Maeurer M. Inflammation and tuberculosis: host-directed therapies. J Intern Med. 2014. doi:10.1111/joim.12256.
Meintjes G, Rangaka MX, Maartens G, Rebe K, Morroni C, Pepper DJ, Wilkinson KA, Wilkinson RJ. Novel relationship between tuberculosis immune reconstitution inflammatory syndrome and antitubercular drug resistance. Clin Infect Dis. 2009;48(5):667–76.
Pasipanodya JG, McNabb SJ, Hilsenrath P, Bae S, Lykens K, Vecino E, Munguia G, Miller TL, Drewyer G, Weis SE. Pulmonary impairment after tuberculosis and its contribution to TB burden. BMC Public Health. 2010;10:259.
Zumla A, Chakaya J, Centis R, D'Ambrosio L, Mwaba P, Bates M, Kapata N, Nyirenda T, Chanda D, Mfinanga S, et al. Tuberculosis treatment and management--an update on treatment regimens, trials, new drugs, and adjunct therapies. Lancet Respir Med. 2015;3(3):220–34.
Pinho ST, Rodrigues P, Andrade RF, Serra H, Lopes JS, Gomes MG. Impact of tuberculosis treatment length and adherence under different transmission intensities. Theor Popul Biol. 2015;104:68–77.
Hoger S, Lykens K, Beavers SF, Katz D, Miller TL. Longevity loss among cured tuberculosis patients and the potential value of prevention. Int J Tuberc Lung Dis. 2014;18(11):1347–52.
Kaufmann SH, Lange C, Rao M, Balaji KN, Lotze M, Schito M, Zumla AI, Maeurer M. Progress in tuberculosis vaccine development and host-directed therapies--a state of the art review. Lancet Respir Med. 2014;2(4):301–20.
Ahmed RK, Rohava Z, Balaji KN, Hoffner SE, Gaines H, Magalhaes I, Zumla A, Skrahina A, Maeurer MJ. Pattern recognition and cellular immune responses to novel Mycobacterium tuberculosis-antigens in individuals from Belarus. BMC Infect Dis. 2012;12(1):41.
Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466(7309):973–7.
Wallis RS, Kim P, Cole S, Hanna D, Andrade BB, Maeurer M, Schito M, Zumla A. Tuberculosis biomarkers discovery: developments, needs, and challenges. Lancet Infect Dis. 2013;13(4):362–72.
Zumla AI, Schito M, Maeurer M. Advancing the portfolio of tuberculosis diagnostics, drugs, biomarkers, and vaccines. Lancet Infect Dis. 2014;14(4):267–9.
Mahon RN, Hafner R. Immune Cell Regulatory Pathways Unexplored as Host-Directed Therapeutic Targets for Mycobacterium tuberculosis: An Opportunity to Apply Precision Medicine Innovations to Infectious Diseases. Clin Infect Dis. 2015;61 suppl 3:S200–16.
Zumla A, Chakaya J, Hoelscher M, Ntoumi F, Rustomjee R, Vilaplana C, Yeboah-Manu D, Rasolof V, Munderi P, Singh N, et al. Towards host-directed therapies for tuberculosis. Nat Rev Drug Discov. 2015. doi:10.1038/nrd4696.
Blum CA, Nigro N, Briel M, Schuetz P, Ullmer E, Suter-Widmer I, Winzeler B, Bingisser R, Elsaesser H, Drozdov D, et al. Adjunct prednisone therapy for patients with community-acquired pneumonia: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet. 2015. doi:10.1016/S0140-6736(14)62447-8.
Remmelts HH, Meijvis SC, Biesma DH, van Velzen-Blad H, Voorn GP, Grutters JC, Bos WJ, Rijkers GT. Dexamethasone downregulates the systemic cytokine response in patients with community-acquired pneumonia. Clin Vaccine Immunol. 2012;19(9):1532–8.
Muthuswamy P, Hu TC, Carasso B, Antonio M, Dandamudi N. Prednisone as adjunctive therapy in the management of pulmonary tuberculosis. Report of 12 cases and review of the literature. Chest. 1995;107(6):1621–30.
Tobin DM, Ramakrishnan L. TB: the Yin and Yang of lipid mediators. Curr Opin Pharmacol. 2013;13(4):641–5.
Zelasko S, Arnold WR, Das A. Endocannabinoid metabolism by cytochrome P450 monooxygenases. Prostaglandins Other Lipid Mediat. 2014. doi:10.1016/j.prostaglandins.2014.11.002.
Agarwal S, Reddy GV, Reddanna P. Eicosanoids in inflammation and cancer: the role of COX-2. Expert Rev Clin Immunol. 2009;5(2):145–65.
Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, Ko DC, Zou Y, Bang ND, Chau TT, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148(3):434–46.
Vilaplana C, Marzo E, Tapia G, Diaz J, Garcia V, Cardona PJ. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J Infect Dis. 2013;208(2):199–202.
Guzman JD, Evangelopoulos D, Gupta A, Birchall K, Mwaigwisya S, Saxty B, McHugh TD, Gibbons S, Malkinson J, Bhakta S. Antitubercular specific activity of ibuprofen and the other 2-arylpropanoic acids using the HT-SPOTi whole-cell phenotypic assay. BMJ Open. 2013;3(6). doi:10.1136/bmjopen-2013-002672.
Berger W, De Chandt MT, Cairns CB. Zileuton: clinical implications of 5-Lipoxygenase inhibition in severe airway disease. Int J Clin Pract. 2007;61(4):663–76.
Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014;511(7507):99–103.
Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. 2005;4(12):977–87.
Kwak B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognized type of immunomodulator. Nat Med. 2000;6(12):1399–402.
Thomsen RW, Riis A, Kornum JB, Christensen S, Johnsen SP, Sorensen HT. Preadmission use of statins and outcomes after hospitalization with pneumonia: population-based cohort study of 29,900 patients. Arch Intern Med. 2008;168(19):2081–7.
Thomsen RW, Hundborg HH, Johnsen SP, Pedersen L, Sorensen HT, Schonheyder HC, Lervang HH. Statin use and mortality within 180 days after bacteremia: a population-based cohort study. Crit Care Med. 2006;34(4):1080–6.
Chalmers JD, Singanayagam A, Murray MP, Hill AT. Prior statin use is associated with improved outcomes in community-acquired pneumonia. Am J Med. 2008;121(11):1002–7. e1001.
Mortensen EM, Restrepo MI, Anzueto A, Pugh J. The effect of prior statin use on 30-day mortality for patients hospitalized with community-acquired pneumonia. Respir Res. 2005;6:82.
Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, Suzuki H, Marais AD, Brombacher F. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis. 2014;209(5):754–63.
Skerry C, Pinn ML, Bruiners N, Pine R, Gennaro ML, Karakousis PC. Simvastatin increases the in vivo activity of the first-line tuberculosis regimen. J Antimicrob Chemother. 2014;69(9):2453–7.
Kang YA, Choi NK, Seong JM, Heo EY, Koo BK, Hwang SS, Park BJ, Yim JJ, Lee CH. The effects of statin use on the development of tuberculosis among patients with diabetes mellitus. Int J Tuberc Lung Dis. 2014;18(6):717–24.
Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: From Mechanisms of Action to Therapies. Cell Metab. 2014;20(6):953–66.
Singhal A, Jie L, Kumar P, Hong GS, Leow MK, Paleja B, Tsenova L, Kurepina N, Chen J, Zolezzi F, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med. 2014;6(263):263ra159.
Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A. 2015;112(6):1809–14.
Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103–7.
Napier RJ, Norris BA, Swimm A, Giver CR, Harris WA, Laval J, Napier BA, Patel G, Crump R, Peng Z, et al. Low doses of imatinib induce myelopoiesis and enhance host anti-microbial immunity. PLoS Pathog. 2015;11(3):e1004770.
Napier RJ, Rafi W, Cheruvu M, Powell KR, Zaunbrecher MA, Bornmann W, Salgame P, Shinnick TM, Kalman D. Imatinib-sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe. 2011;10(5):475–85.
Riches JC, Gribben JG. Immunomodulation and immune reconstitution in chronic lymphocytic leukemia. Semin Hematol. 2014;51(3):228–34.
Boddy AV, Yule SM. Metabolism and pharmacokinetics of oxazaphosphorines. Clin Pharmacokinet. 2000;38(4):291–304.
Hall AG, Tilby MJ. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev. 1992;6(3):163–73.
Dimeloe S, Frick C, Fischer M, Gubser PM, Razik L, Bantug GR, Ravon M, Langenkamp A, Hess C. Human regulatory T cells lack the cyclophosphamide-extruding transporter ABCB1 and are more susceptible to cyclophosphamide-induced apoptosis. Eur J Immunol. 2014;44(12):3614–20.
Kanakry CG, Ganguly S, Zahurak M, Bolanos-Meade J, Thoburn C, Perkins B, Fuchs EJ, Jones RJ, Hess AD, Luznik L. Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci Transl Med. 2013;5(211):211ra157.
Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18(8):1254–61.
Zumla A, Maeurer M. Rational development of adjunct immune-based therapies for drug-resistant tuberculosis: hypotheses and experimental designs. J Infect Dis. 2012;205 Suppl 2:S335–339.
Li N, Xie WP, Kong H, Min R, Hu CM, Zhou XB, Lu ZM, Ji XH, Wang H. Enrichment of regulatory T-cells in blood of patients with multidrug-resistant tuberculosis. Int J Tuberc Lung Dis. 2015;19(10):1230–8.
Singh A, Dey AB, Mohan A, Sharma PK, Mitra DK. Foxp3+ regulatory T cells among tuberculosis patients: impact on prognosis and restoration of antigen specific IFN-gamma producing T cells. PLoS One. 2012;7(9):e44728.
Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Adv Cancer Res. 2004;91:137–68.
Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27(32):5459–68.
Hymes KB. The role of histone deacetylase inhibitors in the treatment of patients with cutaneous T-cell lymphoma. Clin Lymphoma Myeloma Leuk. 2010;10(2):98–109.
Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, Perez-Cardenas E, de la Cruz-Hernandez E, Herrera LA. Valproic acid as epigenetic cancer drug: preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat Rev. 2008;34(3):206–22.
Cang S, Ma Y, Liu D. New clinical developments in histone deacetylase inhibitors for epigenetic therapy of cancer. J Hematol Oncol. 2009;2:22.
Han BR, You BR, Park WH. Valproic acid inhibits the growth of HeLa cervical cancer cells via caspase-dependent apoptosis. Oncol Rep. 2013. doi:10.3892/or.2013.2747.
Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro Oncol. 2010;12(4):328–40.
Campbell GR, Bruckman RS, Chu YL, Spector SA. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. J Biol Chem. 2015;290(8):5028–40.
Ylisastigui L, Archin NM, Lehrman G, Bosch RJ, Margolis DM. Coaxing HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. Aids. 2004;18(8):1101–8.
Archin NM, Keedy KS, Espeseth A, Dang H, Hazuda DJ, Margolis DM. Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. Aids. 2009;23(14):1799–806.
Yang HC, Xing S, Shan L, O'Connell K, Dinoso J, Shen A, Zhou Y, Shrum CK, Han Y, Liu JO, et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest. 2009;119(11):3473–86.
Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, Zhang H, Margolick JB, Blankson JN, Siliciano RF. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012;36(3):491–501.
Rasmussen TA, Tolstrup M, Winckelmann A, Ostergaard L, Sogaard OS. Eliminating the latent HIV reservoir by reactivation strategies: Advancing to clinical trials. Hum Vaccin Immunother. 2013;9(4):790–9.
Shirakawa K, Chavez L, Hakre S, Calvanese V, Verdin E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 2013;21(6):277–85.
Nelson J, Roe K, Orillo B, Shi PY, Verma S. Combined treatment of adenosine nucleoside inhibitor NITD008 and histone deacetylase inhibitor vorinostat represents an immunotherapy strategy to ameliorate West Nile virus infection. Antiviral Res. 2015;122:39–45.
Coussens AK, Wilkinson RJ, Martineau AR. Phenylbutyrate Is Bacteriostatic against Mycobacterium tuberculosis and Regulates the Macrophage Response to Infection, Synergistically with 25-Hydroxy-Vitamin D(3). PLoS Pathog. 2015;11(7):e1005007.
Mily A, Rekha RS, Kamal SM, Arifuzzaman AS, Rahim Z, Khan L, Haq MA, Zaman K, Bergman P, Brighenti S, et al. Significant Effects of Oral Phenylbutyrate and Vitamin D3 Adjunctive Therapy in Pulmonary Tuberculosis: A Randomized Controlled Trial. PLoS One. 2015;10(9):e0138340.
Wallis RS, van Vuuren C, Potgieter S. Adalimumab treatment of life-threatening tuberculosis. Clin Infect Dis. 2009;48(10):1429–32.
Casarini M, Ameglio F, Alemanno L, Zangrilli P, Mattia P, Paone G, Bisetti A, Giosue S. Cytokine levels correlate with a radiologic score in active pulmonary tuberculosis. Am J Respir Crit Care Med. 1999;159(1):143–8.
Chowdhury IH, Ahmed AM, Choudhuri S, Sen A, Hazra A, Pal NK, Bhattacharya B, Bahar B. Alteration of serum inflammatory cytokines in active pulmonary tuberculosis following anti-tuberculosis drug therapy. Mol Immunol. 2014;62(1):159–68.
Okada M, Kita Y, Kanamaru N, Hashimoto S, Uchiyama Y, Mihara M, Inoue Y, Ohsugi Y, Kishimoto T, Sakatani M. Anti-IL-6 receptor antibody causes less promotion of tuberculosis infection than anti-TNF-alpha antibody in mice. Clin Dev Immunol. 2011;2011:404929.
Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, Nakano N, Ikeda Y, Sasaki T, Nishioka K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627–32.
van Rhee F, Wong RS, Munshi N, Rossi JF, Ke XY, Fossa A, Simpson D, Capra M, Liu T, Hsieh RK, et al. Siltuximab for multicentric Castleman's disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15(9):966–74.
Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012;38(7):904–10.
Datta M, Via LE, Kamoun WS, Liu C, Chen W, Seano G, Weiner DM, Schimel D, England K, Martin JD, et al. Anti-vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proc Natl Acad Sci U S A. 2015;112(6):1827–32.
Oehlers SH, Cronan MR, Scott NR, Thomas MI, Okuda KS, Walton EM, Beerman RW, Crosier PS, Tobin DM. Interception of host angiogenic signalling limits mycobacterial growth. Nature. 2015;517(7536):612–5.
Veenstra H, Baumann R, Lukey PT, Beyers N, van Helden PD, Walzl G. High levels of intracellular IL-4 are expressed in circulating apoptotic T cells in patients with tuberculosis and in community controls. Tuberculosis. 2008;88(1):21–30.
Roberts T, Beyers N, Aguirre A, Walzl G. Immunosuppression during active tuberculosis is characterized by decreased interferon- gamma production and CD25 expression with elevated forkhead box P3, transforming growth factor- beta, and interleukin-4 mRNA levels. J Infect Dis. 2007;195(6):870–8.
Heitmann L, Abad Dar M, Schreiber T, Erdmann H, Behrends J, McKenzie AN, Brombacher F, Ehlers S, Holscher C. The IL-13/IL-4Ralpha axis is involved in tuberculosis-associated pathology. J Pathol. 2014;234(3):338–50.
Chen H, Cheng C, Li M, Gao S, Li S, Sun H. Expression of TNF-alpha, IFN-gamma, TGF-beta, and IL-4 in the spinal tuberculous focus and its impact on the disease. Cell Biochem Biophys. 2014;70(3):1759–64.
Rahman S, Rehn A, Rahman J, Andersson J, Svensson M, Brighenti S. Pulmonary tuberculosis patients with a vitamin D deficiency demonstrate low local expression of the antimicrobial peptide LL-37 but enhanced FoxP3 regulatory T cells and IgG-secreting cells. Clin Immunol. 2014;156(2):85–97.
Montoya D, Inkeles MS, Liu PT, Realegeno S, Teles RM, Vaidya P, Munoz MA, Schenk M, Swindell WR, Chun R, et al. IL-32 is a molecular marker of a host defense network in human tuberculosis. Sci Transl Med. 2014;6(250):250ra114.
Klug-Micu GM, Stenger S, Sommer A, Liu PT, Krutzik SR, Modlin RL, Fabri M. CD40 ligand and interferon-gamma induce an antimicrobial response against Mycobacterium tuberculosis in human monocytes. Immunology. 2013;139(1):121–8.
Daley P, Jagannathan V, John KR, Sarojini J, Latha A, Vieth R, Suzana S, Jeyaseelan L, Christopher DJ, Smieja M, et al. Adjunctive vitamin D for treatment of active tuberculosis in India: a randomised, double-blind, placebo-controlled trial. Lancet Infect Dis. 2015;15(5):528–34.
Tukvadze N, Sanikidze E, Kipiani M, Hebbar G, Easley KA, Shenvi N, Kempker RR, Frediani JK, Mirtskhulava V, Alvarez JA, et al. High-dose vitamin D3 in adults with pulmonary tuberculosis: a double-blind randomized controlled trial. Am J Clin Nutr. 2015;102(5):1059–69.
Azoury SC, Straughan DM, Shukla V. Immune Checkpoint Inhibitors for Cancer Therapy: Clinical Efficacy and Safety. Curr Cancer Drug Targets. 2015;15(6):452–62.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Leung J, Suh WK. The CD28-B7 Family in Anti-Tumor Immunity: Emerging Concepts in Cancer Immunotherapy. Immune Netw. 2014;14(6):265–76.
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42.
Srahna M, Van Grunsven LA, Remacle JE, Vandenberghe P. CTLA-4 interacts with STAT5 and inhibits STAT5-mediated transcription. Immunology. 2006;117(3):396–401.
Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3 - potential mechanisms of action. Nat Rev Immunol. 2014;15(1):45–56.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61.
Gardiner D, Lalezari J, Lawitz E, DiMicco M, Ghalib R, Reddy KR, Chang KM, Sulkowski M, Marro SO, Anderson J, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS One. 2013;8(5):e63818.
Jurado JO, Alvarez IB, Pasquinelli V, Martinez GJ, Quiroga MF, Abbate E, Musella RM, Chuluyan HE, Garcia VE. Programmed death (PD)-1:PD-ligand 1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J Immunol. 2008;181(1):116–25.
Singh A, Mohan A, Dey AB, Mitra DK. Inhibiting the programmed death 1 pathway rescues Mycobacterium tuberculosis-specific interferon gamma-producing T cells from apoptosis in patients with pulmonary tuberculosis. J Infect Dis. 2013;208(4):603–15.
Hassan SS, Akram M, King EC, Dockrell HM, Cliff JM. PD-1, PD-L1 and PD-L2 Gene Expression on T-Cells and Natural Killer Cells Declines in Conjunction with a Reduction in PD-1 Protein during the Intensive Phase of Tuberculosis Treatment. PLoS One. 2015;10(9):e0137646.
McNab FW, Berry MP, Graham CM, Bloch SA, Oni T, Wilkinson KA, Wilkinson RJ, Kon OM, Banchereau J, Chaussabel D, et al. Programmed death ligand 1 is over-expressed by neutrophils in the blood of patients with active tuberculosis. Eur J Immunol. 2011;41(7):1941–7.
Singh A, Dey AB, Mohan A, Mitra DK. Programmed death-1 receptor suppresses gamma-IFN producing NKT cells in human tuberculosis. Tuberculosis. 2014;94(3):197–206.
Garcia Jacobo RE, Serrano CJ, Enciso Moreno JA, Gaspar Ramirez O, Trujillo Ochoa JL, Uresti Rivera EE, Portales Perez DP, Gonzalez-Amaro R, Garcia Hernandez MH. Analysis of Th1, Th17 and regulatory T cells in tuberculosis case contacts. Cell Immunol. 2014;289(1-2):167–73.
Merlo A, Saverino D, Tenca C, Grossi CE, Bruno S, Ciccone E. CD85/LIR-1/ILT2 and CD152 (cytotoxic T lymphocyte antigen 4) inhibitory molecules down-regulate the cytolytic activity of human CD4+ T-cell clones specific for Mycobacterium tuberculosis. Infect Immun. 2001;69(10):6022–9.
Sierro S, Romero P, Speiser DE. The CD4-like molecule LAG-3, biology and therapeutic applications. Expert Opin Ther Targets. 2011;15(1):91–101.
Phillips BL, Mehra S, Ahsan MH, Selman M, Khader SA, Kaushal D. LAG3 Expression in Active Mycobacterium tuberculosis Infections. Am J Pathol. 2014. doi:10.1016/j.ajpath.2014.11.003.
Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, Waldschmidt TJ, Crompton PD, Harty JT. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. 2012;13(2):188–95.
Ngiow SF, Teng MW, Smyth MJ. Prospects for TIM3-Targeted Antitumor Immunotherapy. Cancer Res. 2011;71(21):6567–71.
Sada-Ovalle I, Chavez-Galan L, Torre-Bouscoulet L, Nava-Gamino L, Barrera L, Jayaraman P, Torres-Rojas M, Salazar-Lezama MA, Behar SM. The Tim3-galectin 9 pathway induces antibacterial activity in human macrophages infected with Mycobacterium tuberculosis. J Immunol. 2012;189(12):5896–902.
Sada-Ovalle I, Ocana-Guzman R, Perez-Patrigeon S, Chavez-Galan L, Sierra-Madero J, Torre-Bouscoulet L, Addo MM. Tim-3 blocking rescue macrophage and T cell function against Mycobacterium tuberculosis infection in HIV+ patients. J Int AIDS Soc. 2015;18(1):20078.
Parida SK, Poiret T, Zhenjiang L, Meng Q, Heyckendorf J, Lange C, Ambati AS, Rao MV, Valentini D, Ferrara G, et al. T-Cell Therapy: Options for Infectious Diseases. Clin Infect Dis. 2015;61 suppl 3:S217–24.
Parida SK, Madansein R, Singh N, Padayatchi N, Master I, Naidu K, Zumla A, Maeurer M. Cellular therapy in tuberculosis. Int J Infect Dis. 2015;32:32–8.
Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–8.
Skrahin A, Ahmed RK, Ferrara G, Rane L, Poiret T, Isaikina Y, Skrahina A, Zumla A, Maeurer MJ. Autologous mesenchymal stromal cell infusion as adjunct treatment in patients with multidrug and extensively drug-resistant tuberculosis: an open-label phase 1 safety trial. Lancet Respir Med. 2014;2(2):108–22.
Cho S, Mehra V, Thoma-Uszynski S, Stenger S, Serbina N, Mazzaccaro RJ, Flynn JL, Barnes PF, Southwood S, Celis E, et al. Antimicrobial activity of MHC class I-restricted CD8+ T cells in human tuberculosis. Proc Natl Acad Sci U S A. 2000;97(22):12210–5.
Mustafa AS, Oftung F, Amoudy HA, Madi NM, Abal AT, Shaban F, Rosen Krands I, Andersen P. Multiple epitopes from the Mycobacterium tuberculosis ESAT-6 antigen are recognized by antigen-specific human T cell lines. Clin Infect Dis. 2000;30 Suppl 3:S201–205.
Kasmar AG, Van Rhijn I, Magalhaes KG, Young DC, Cheng TY, Turner MT, Schiefner A, Kalathur RC, Wilson IA, Bhati M, et al. Cutting Edge: CD1a tetramers and dextramers identify human lipopeptide-specific T cells ex vivo. J Immunol. 2013;191(9):4499–503.
Axelsson-Robertson R, Rao M, Loxton AG, Walzl G, Bates M, Zumla A, Maeurer M. Frequency of Mycobacterium tuberculosis-specific CD8+ T-cells in the course of anti-tuberculosis treatment. Int J Infect Dis. 2015;32:23–9.
El Daker S, Sacchi A, Montesano C, Altieri AM, Galluccio G, Colizzi V, Martini F, Martino A. An abnormal phenotype of lung Vgamma9Vdelta2 T cells impairs their responsiveness in tuberculosis patients. Cell Immunol. 2013;282(2):106–12.
Beeson CC, Beeson GC, Schnellmann RG. A high-throughput respirometric assay for mitochondrial biogenesis and toxicity. Anal Biochem. 2010;404(1):75–81.
Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153(3):521–34.
Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, Landay AL, Coombs RW, Richman DD, Mellors JW, et al. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet. 2005;366(9485):549–55.
Tung EW, Winn LM. Valproic acid increases formation of reactive oxygen species and induces apoptosis in postimplantation embryos: a role for oxidative stress in valproic acid-induced neural tube defects. Mol Pharmacol. 2011;80(6):979–87.
Schiebler M, Brown K, Hegyi K, Newton SM, Renna M, Hepburn L, Klapholz C, Coulter S, Obregon-Henao A, Henao Tamayo M, et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol Med. 2015;7(2):127–39.
Sung JA, Lam S, Garrido C, Archin N, Rooney CM, Bollard CM, Margolis DM. Expanded Cytotoxic T-cell Lymphocytes Target the Latent HIV Reservoir. J Infect Dis. 2015. doi:10.1093/infdis/jiv022.
Petruccelli LA, Dupere-Richer D, Pettersson F, Retrouvey H, Skoulikas S, Miller Jr WH. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One. 2011;6(6):e20987.
Marks PA, Richon VM, Kelly WK, Chiao JH, Miller T. Histone deacetylase inhibitors: development as cancer therapy. Novartis Found Symp. 2004;259:269–81. discussion 281-268.
Kulkarni NN, Yi Z, Huehnken C, Agerberth B, Gudmundsson GH. Phenylbutyrate induces cathelicidin expression via the vitamin D receptor: Linkage to inflammatory and growth factor cytokines pathways. Mol Immunol. 2015;63(2):530–9.
Zhao J, Cao Y, Lei Z, Yang Z, Zhang B, Huang B. Selective depletion of CD4 + CD25 + Foxp3+ regulatory T cells by low-dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer Res. 2010;70(12):4850–8.
Henter JI, Palmkvist-Kaijser K, Holzgraefe B, Bryceson YT, Palmer K. Cytotoxic therapy for severe swine flu A/H1N1. Lancet. 2010;376(9758):2116.
Pirinen E, Canto C, Jo YS, Morato L, Zhang H, Menzies KJ, Williams EG, Mouchiroud L, Moullan N, Hagberg C, et al. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014;19(6):1034–41.
Zumla A, Bates M, Maeurer M. Corticosteroid therapy for pneumonia. Lancet. 2015;386(9997):955.
Singh NP, Hegde VL, Hofseth LJ, Nagarkatti M, Nagarkatti P. Resveratrol (trans-3,5,4′-trihydroxystilbene) ameliorates experimental allergic encephalomyelitis, primarily via induction of apoptosis in T cells involving activation of aryl hydrocarbon receptor and estrogen receptor. Mol Pharmacol. 2007;72(6):1508–21.
Estrella JL, Kan-Sutton C, Gong X, Rajagopalan M, Lewis DE, Hunter RL, Eissa NT, Jagannath C. A Novel in vitro Human Macrophage Model to Study the Persistence of Mycobacterium tuberculosis Using Vitamin D(3) and Retinoic Acid Activated THP-1 Macrophages. Front Microbiol. 2011;2:67.
van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68–78.
Oh S, Perera LP, Terabe M, Ni L, Waldmann TA, Berzofsky JA. IL-15 as a mediator of CD4+ help for CD8+ T cell longevity and avoidance of TRAIL-mediated apoptosis. Proc Natl Acad Sci U S A. 2008;105(13):5201–6.
Borch TH, Donia M, Andersen MH, Svane IM. Reorienting the immune system in the treatment of cancer by using anti-PD-1 and anti-PD-L1 antibodies. Drug Discov Today. 2015. doi:10.1016/j.drudis.2015.07.003.
Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, Powderly JD, Heist RS, Carvajal RD, Jackman DM, et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2015;33(18):2004–12.
Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–59.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71.
Niezgoda A, Niezgoda P, Czajkowski R. Novel Approaches to Treatment of Advanced Melanoma: A Review on Targeted Therapy and Immunotherapy. BioMed Res Int. 2015;2015:851387.
Goovaerts O, Jennes W, Massinga-Loembe M, Ceulemans A, Worodria W, Mayanja-Kizza H, Colebunders R, Kestens L, Group T-IS. LPS-binding protein and IL-6 mark paradoxical tuberculosis immune reconstitution inflammatory syndrome in HIV patients. PLoS One. 2013;8(11):e81856.
Rossi JF, Lu ZY, Jourdan M, Klein B. Interleukin-6 as a therapeutic target. Clin Cancer Res. 2015;21(6):1248–57.
Lambrechts D, Lenz HJ, de Haas S, Carmeliet P, Scherer SJ. Markers of response for the antiangiogenic agent bevacizumab. J Clin Oncol. 2013;31(9):1219–30.
Joshi L, Chelluri LK, Gaddam S. Mesenchymal Stromal Cell Therapy in MDR/XDR Tuberculosis: A Concise Review. Arch Immunol Ther Exp (Warsz). 2015. doi:10.1007/s00005-015-0347-9.
Axelsson-Robertson R, Loxton AG, Walzl G, Ehlers MM, Kock MM, Zumla A, Maeurer M. A Broad Profile of Co-Dominant Epitopes Shapes the Peripheral Mycobacterium tuberculosis Specific CD8+ T-Cell Immune Response in South African Patients with Active Tuberculosis. PLoS One. 2013;8(3):e58309.
ᅟ
ᅟ
Authors’ contributions
All authors contributed equally to this review. All authors read and approved the final manuscript.
Authors’ information
AZ is Professor of Infectious Diseases and International Health at the University College London and Consultant Infectious Diseases Physician at the UCL Hospitals NHS Foundation Trust. MR is a postdoctoral scientist in the Therapeutic Immunology Unit (TIM) at the Department of Laboratory Medicine (LABMED), Karolinska Institutet, Sweden. ED is the chief of neurosurgery at Karolinska University Hospital (Solna campus), and adjunct professor at Karolinska Institutet, Sweden. MM is Professor of Clinical Immunology and Head of TIM LABMED, Karolinska Institutet, Sweden.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zumla, A., Rao, M., Dodoo, E. et al. Potential of immunomodulatory agents as adjunct host-directed therapies for multidrug-resistant tuberculosis. BMC Med 14, 89 (2016). https://doi.org/10.1186/s12916-016-0635-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-016-0635-1