
Abstract
Background
Cultivated hexaploid oat (Common oat; Avena sativa) has held a significant place within the global crop community for centuries; although its cultivation has decreased over the past century, its nutritional benefits have garnered increased interest for human consumption. We report the development of fully annotated, chromosome-scale assemblies for the extant progenitor species of the As- and Cp-subgenomes, Avena atlantica and Avena eriantha respectively. The diploid Avena species serve as important genetic resources for improving common oat’s adaptive and food quality characteristics.
Results
The A. atlantica and A. eriantha genome assemblies span 3.69 and 3.78 Gb with an N50 of 513 and 535 Mb, respectively. Annotation of the genomes, using sequenced transcriptomes, identified ~ 50,000 gene models in each species—including 2965 resistance gene analogs across both species. Analysis of these assemblies classified much of each genome as repetitive sequence (~ 83%), including species-specific, centromeric-specific, and telomeric-specific repeats. LTR retrotransposons make up most of the classified elements. Genome-wide syntenic comparisons with other members of the Pooideae revealed orthologous relationships, while comparisons with genetic maps from common oat clarified subgenome origins for each of the 21 hexaploid linkage groups. The utility of the diploid genomes was demonstrated by identifying putative candidate genes for flowering time (HD3A) and crown rust resistance (Pc91). We also investigate the phylogenetic relationships among other A- and C-genome Avena species.
Conclusions
The genomes we report here are the first chromosome-scale assemblies for the tribe Poeae, subtribe Aveninae. Our analyses provide important insight into the evolution and complexity of common hexaploid oat, including subgenome origin, homoeologous relationships, and major intra- and intergenomic rearrangements. They also provide the annotation framework needed to accelerate gene discovery and plant breeding.
Similar content being viewed by others

Background
Oat (Avena sativa L.) is a nutritionally important crop throughout the world. It is ranked 6th in world cereal production [1], and while its primary use continues to be as a livestock feed, its uses as a human food and for cosmetics continue to increase [2]. Among the many nutritional benefits of oat are its high levels of calcium, β-glucan soluble fiber [3,4,5,6], and high-quality oil and protein [7, 8]. Oat seed contains no gluten and only low levels of gluten-related prolamins and therefore is a healthy diet alternative for many people who cannot tolerate dietary gluten. Oat is high in polyphenolic avenanthramides having antioxidant, anti-inflammatory, and antiatherogenic properties [9]. Oat also contains two classes of saponins: avenacosides (sugars bound to steroids) and avenacins (sugars bound to triterpenoid), both of which have been shown to lower cholesterol, stimulate the immune system, and have anti-carcinogenic properties [7]. Oat also has many topical uses, as it has a soothing effect on skin and has been used to treat dry, itchy skin [10]; oat has also been shown to have sun-blocking properties [11], and it is often found in products to treat eczema, psoriasis, and other skin conditions [12, 13].
Common oat (A. sativa) and red oat (A. byzantina C. Koch) are allohexaploids (2n = 6x = 42, AACCDD subgenomes) belonging to the Poeae Tribe of the Poaceae [14] and are thought to have been domesticated from wild-weedy A. sterilis L. [15], a species that arose from hybridization between a CCDD allotetraploid closely related to modern A. insularis Ladiz. and an AsAs diploid [16]. Several variants of the A-subgenome diploids exist (Ac, Ad, Al, Ap, and As [17];) and are known to harbor several genetic features of significance, including major crown rust resistance genes that have been transferred into hexaploid oat cultivars [18, 19]. The A-genome diploids have also been identified as potential gene sources for improving soluble fiber and protein [20]. The A-subgenome is also part of a major intergenomic translocation (7C-17A) in A. sativa-A. sterilis that has been associated with adaptation to winter hardiness—key elements in oat production that likely contributed to the plant’s ability to shift from Mediterranean winter ecology to Eurasian spring-summer cultivation [21].
The C-subgenome chromosomes have a high amount of diffuse heterochromatin along their entirety [22]; this is a genetic feature not seen in the A and D chromosomes, where heterochromatin is localized and seemingly concentrated around the centromeres, at the telomeres, and in flanking secondary constrictions where rRNA genes are located. Among the important genetic features in the C-subgenome is a terminal translocation segment on the long arm of 21D which carries a putative CSlF6c locus that likely has a negative effect on seed soluble fiber content [23, 24]. Linkage has also been demonstrated between the chromosome 5C telomeric knob in allotetraploid A. magna Murphy et Terrell (CCDD subgenomes) and co-segregating genes controlling awn production and basal abscission layer formation which have been implicated in the domestication of common oat [25].
Despite the historical importance of oat and the renewed interest in its nutritional value, a complete genome sequence of oat has yet to be reported. Indeed, the A. sativa genome is large (> 12 Gb [26]), duplicated, complex, highly repetitive, and characterized by several major intra- and intergenomic rearrangements—making full genome assembly of the hexaploid difficult [27]. Here we report the development of fully annotated, chromosome-scale assemblies for the extant progenitor species of the As- and Cp-subgenomes, A. atlantica B.R.Baum & Fedak and Avena eriantha Durieu., respectively. Using these assemblies, we (i) identified and quantified repetitive element content in the genome, including centromeric and telomeric repeats, (ii) analyzed syntenic relationships with other cereal grains and homoeologous relationships within oat using consensus linkage maps [28], (iii) identified putative candidate genes for flowering time [29] and crown rust resistance [30] relative to recently published genome-wide association studies (GWAS), (iv) estimated the age of the evolutionary split between the A- and C-subgenomes using synonymous substitution rates (Ks) analysis, and (v) examined the genetic diversity and phylogenetic relationship from a resequencing panel of 76 A- and C-genome Avena species.
Results
Whole-genome sequencing and assembly
We selected the A. atlantica accession Cc 7277 and the A. eriantha accession CN 19328 for whole-genome shotgun sequencing. Both accessions are highly inbred and phenotypically homogeneous and represent type accessions for their respective species. A total of 31,544,396 and 28,257,346 PacBio reads were generated across 122 (RSII and Sequel) and 54 (Sequel) SMRT cells generating a total of 325.9 (~84× coverage) and 276.6 (~71× coverage) Gb of sequence data for A. atlantica and A. eriantha, respectively. The longest reads for each species, 194,884 and 151,576 bp, came from the Sequel instrument. The N50 read length for A. atlantica and A. eriantha was 18,658 and 15,102 bp, respectively. In addition to PacBio sequencing, a total of 192 Gb for A. atlantica and 40 Gb for A. eriantha of 2 × 150 bp Illumina sequences were generated. A k-mer analysis (at k = 21 scale) using Genoscope [31] predicted a genome size of 3.72 Gb with 0.07% heterozygosity and a repeat fraction of 78% for A. atlantica and a genome size of 4.17 Gb with a 0.12% heterozygosity and a repeat fraction of 76% for A. eriantha. The relative magnitude of these values agree well with those reported by Bennett and Smith [26] and Yan et al. [32], both of which report that the genomes of the A-genome diploids are ~ 15% smaller than the C-genome diploids. However, former estimates determined by replicated flow cytometry measurements ranged in size from 4.1 to 4.6 Gb for A-genomes and from 5.0 to 5.1 Gb for C-genomes [32]. The differences in genome size predicted by k-mer vs. flow cytometry analyses are likely a reflection of the significant repeat fraction in the oat genome that is difficult to account for using a k-mer analysis.
Prior to Hi-C scaffolding, Canu was used to assemble the A. atlantica and A. eriantha PacBio long reads into 3914 and 8067 contigs with an N50 of 5,544,947 and 1,385,002 bp, spanning a total of 3.68 and 3.77 Gb of total length, respectively (Table 1). The L50 of the assemblies were 196 and 797 and the longest contigs spanned 25,143,700 and 10,103,775 bp, respectively. The average G+C content of the assemblies were 44.4% and 43.9%, which is similar to most monocotyledonous cereals (e.g., Sorghum bicolor, 43.9%; Oryza sativa, 43.6% G+C) but significantly higher than G+C content predicted for dicots, which typically range between 33 and 36% (e.g., Carica papaya, 34%; Arabidopsis thaliana, 36%) [34]. As these were PacBio read based assemblies, no “N” gaps were present in the Canu assemblies.
To improve the Canu assemblies, contigs were further scaffolded by chromatin-contact maps using DoveTail Chicago® and Hi-C libraries. Chicago® library contact maps are based on purified DNA that is reconstituted in vitro and thus limited to chromatin associations no larger than the size of the purified input DNA fragments (< 100 kb). Nonetheless, they are ideal for detecting and correcting miss-joins in de novo assemblies as well as short-range scaffolding [35]. Approximately 73× coverage of 1–100 kb read pairs (2 × 150) were generated from Chicago® libraries for each Avena species and used to scaffold the Canu assemblies using the HiRiSE™ scaffolder. In total, 334 and 158 breaks were made, while 1157 and 2962 joins were made in the A. atlantica and A. eriantha assemblies, respectively. The net effect of these changes was to decrease the number of total scaffolds to 3118 in the A. atlantica assembly and to 5263 in the A. eriantha assembly, which was accompanied by a slight decrease in N50 (4310 and 1.314 kb, respectively) for each assembly. Whenever a join was made between contigs, an “N” gap, consisting of 100 Ns, was created. The total percent of the genome in gaps, for both species, was less than 0.1%.
The Chicago®-based assemblies were then further scaffolded using in vivo Hi-C libraries, created from native chromatin to produce ultra-long-range mate pairs. Mate pair reads (10–10,000 kb) representing a physical coverage of 2749× and 513× were generated for the A. atlantica and A. eriantha genomes and scaffolded using the HiRiSE™ scaffolder. In total, 922 joins and 2614 joins (plus three breaks) in the A. atlantica and A. eriantha were made, respectively, producing ultra-long scaffolds, putatively representing full-length chromosomes and/or chromosome arms. The HiRiSE™ assembly for A. atlantica had a scaffold N50 of 513.2 Mb, and an L50 of 4, spanning a total sequence length of 3.685 Gb. The longest scaffold spanned 577.8 Mb. Similarly, the A. eriantha assembly had a scaffold N50 of 534.8 Mb, an L50 of 4, and spanned a total sequence length of 3.778 Gb with the longest scaffold reaching 588.3 Mb. Scaffold joins produced by the Hi-C mate pairs introduced new “N” gaps in the assembly (each consisting of 1000 Ns), thereby increasing the number of gaps in the assembly to 2079 and 5576 for A. atlantica and A. eriantha, respectively. The final percentage of “N” nucleotides in the final assemblies was less than 0.1%, with the average gap size of 600 and 578 bp, respectively (Table 1).
The longest eight scaffolds of the A. atlantica assembly, presumably representing two chromosome arms (205 and 278 Mb) and six full-length chromosomes (448–577 Mb), consisted of > 96% of the total sequence length from the Canu assembly. Similarly, the longest seven scaffolds, ranging in size from 455 to 588 Mb, presumably representing each of the seven haploid A. eriantha chromosomes, were composed of > 97% of the total Canu assembly sequence. For simplicity, scaffolds representing each of the seven chromosomes from each species are referred to forthwith by size (longest to shortest) as AA1-AA7 and AE1-AE7. The scaffolds in the A. atlantica and A. eriantha assemblies that remain unintegrated into one of the chromosome-scale pseudomolecules are relatively small and repetitive, with an average size of 61 and 38 kb, which likely contributed to the inability of the proximity-guided assembler to confidently place these contigs within the framework of the chromosomes—specifically the low number of interactions on a short fragment as well as the inability to discern interaction distance differences over the short molecule.
Chromosome arm merging
We compared the A. atlantica assembly with a recently published genetic linkage map, constructed from an F6:8 recombinant inbred line population generated from a cross of A. strigosa x A. wiestii, both AsAs Avena diploid species [36]. This map was based on 11,455 ordered, codominant 64-base tag-level haplotypes on seven linkage groups generated using the Haplotag pipeline [37]. Of these, 4551 haplotypes had perfect matches to single sites on the eight largest scaffolds. A clear one-to-one correspondence between linkage groups (LG) and physical assembly scaffolds was observed (Fig. 1), with greater than 97% of the tag-level haplotypes mapping to a specific scaffold derived from a single LG. For example, of the 846 tag-level haplotypes mapping to scaffold ScoFOjO_324_449 (AA1), 838 (> 99%) were derived from LG 7 (Table 2). Of the 464 tag-level haplotypes derived from LG 2, 378 mapped to scaffold ScoFOjO_1310 (278 Mb) and 85 mapped to scaffold ScoFOjO_1577 (205 Mb), indicating that these two smaller scaffolds should be merged to produce a single, full-length pseudo chromosome (AA5; 485 Mb), thus completing the assembly of seven full-length haploid chromosomes for A. atlantica. A head-to-tail merging of these chromosome arms (separated by 1000 Ns) was determined based on the collinearity of the tag-level haplotypes with respect to their orientation within the linkage group. A near perfect collinear relationship was observed between the linkage map and the physical map for all chromosome-linkage group comparisons, with the exceptions being the anticipated reductions of linkage distances relative to physical distances observed at the pericentromeric regions of each chromosome (Fig. 1). It is well documented that recombination is suppressed in centromeres at rates ranging from fivefold to greater than 200-fold, depending on the species [38, 39]. Of the 2188 contigs that were unintegrated into an A. atlantica chromosome using the Hi-C data, we identified segregating haplotypes linked to 22, spanning a total length of 1.07 Mb, which could tentatively place them into the context of the seven haploid chromosomes based on their linkage position (Fig. 1).

Correlation between the physical and linkage map. The genetic position of mapped markers is plotted as a function of physical distance relative to the A. atlantica genome assembly. The linkage position of six unassigned scaffolds with multiple mapping markers is shown
Analysis of repetitive elements
The repeat fraction of the Avena genome assemblies was identified and annotated using RepeatModeler and RepeatMasker. In total, ~ 83% of each genome was classified as repetitive, with the most commonly identified repetitive elements being classified as long terminal repeat retrotransposons (LTR-RTs); LTR-RTs are the most abundant genomic components in flowering plants [40, 41], and their abundance is strongly correlated with genome size [42]. Within published plant genomes, repeat content varies widely, ranging from 3% for the minute 82 Mb genome of Utricularia gibba L. [43] to 85% for maize [44]. Given the large size of these genomes, it is not surprising that < 20% of the genome is classified as non-repetitive.
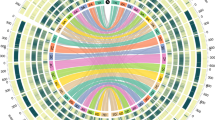
Of the various LTR-RT present (Additional file 1: Table S1), Gypsy-like and Copia-like elements represent > 60% of each genome, in a ratio of 2.3:1 and 3.5:1 for the A. atlantica and A. eriantha genomes, respectively, which is similar to the ratios reported for other Poaceae species (e.g., rice, 4.9:1 [45]; sorghum, 3.7:1 [46]; and maize, 1.6:1, [47]). The next most common element was class II CMC-EnSpm DNA transposons, representing ~ 5% of each genome—which are known common features of the cereals [48, 49]. Interestingly, a significant proportion (A. atlantica: 10.6% and A. eriantha: 14.3%) of the interspersed repeat fraction for each genome was classified as “unknown”. Given the extensive investigations of repeat elements in the grasses [50,51,52], this unknown fraction likely represents repeat elements unique to Avena and could be invaluable in differentiating the A-, C-, and D-subgenomes in hexaploid oat. For example, Solano et al. [53] reported the identification of a tandem repeat sequence clone (pAm1; GenBank X83958) from Avena murphyi L., an AACC tetraploid, which selectively hybridized to the C-subgenome. A repeat that was highly homologous (E-value 2E-82) to pAm1 was identified by RepeatModeler in A. eriantha, but is missing in the A. atlantica genome (Fig. 2a; Tracks 4 and 5). Similarly, Katsiotis et al. [54] reported the identification of an interspersed repeat (pAvKB26; GenBank AJ297385.1) that selectively hybridized to only the A- and D-subgenomes. This repeat was identified in the unknown repeat fraction of A. atlantica but was missing in the A. eriantha genome (Fig. 2b; Tracks 4 and 5). Repeat content is believed to be an important driver of genome organization and evolution [55] and these data will be important for understanding the overall evolution of common hexaploid oat.

Genome overview of a A. atlantica and b A. eriantha. Track 1 (outside): Chromosome and sizes; Tracks 2: Annotated gene density; Track 3: Centromeric repeat density; Track 4: Telomeric sub-repeat density; Track 5: C-genome specific repeat (pAm1) density; Track 6: A-genome specific repeat (pAvKB26) density
In addition to the interspersed repeat elements, ~ 0.5% of the genome was classified as low complexity, satellite, microsatellite or telomeric repeat (see genomic feature section below). Indeed, 5217 and 3404 putative microsatellite loci were identified, with the most common di-, tri- and tetranucleotide repeat motif identified being (AT)n, (AAC)n or (GGC)n and (TTTA)n, in A. atlantica and A. eriantha, respectively. To date, no microsatellites have been generated specifically for the Avena diploid species – thus these new putative microsatellite loci represent important genetic tools for studying diversity and specifically for advancing breeding in the A-genome diploids.
Transcriptome assembly and functional annotation
The A. atlantica and A. eriantha transcriptomes, which consisted of 51,223 and 47,361 scaffolded isoforms, the Brachypodium cDNA and peptide models (v 1.0; Ensembl genomes) and the uniprot-sprot database were provided as primary evidence for annotation in the MAKER pipeline [56]. The RNA-Seq data mapped with high efficiency to the assemblies, with > 96% of the reads mapping to their respective genome at 93.1% concordance for pair alignment rates, suggestive of high-quality genome assemblies for both species. The MAKER pipeline identified a total of 51,100 and 49,105 gene predictions, with mean transcript lengths of 3018 and 3153 bp, and with 70% and 66% of the annotations having annotation edit distance (AED) measures < 0.25, for A. atlantica and A. eriantha genomes, respectively. AED integrates sensitivity, specificity, and accuracy measurement to calculate annotation quality, where AED values < 0.25 are indicative of high-quality annotations [57]. The mean G+C content of the transcripts in both species was ~ 52%. The increase in G+C content within coding regions relative to the overall G+C content of the genome (~ 44%) is a well-known phenomenon and is hypothesized to be the result of GC-biased gene conversion – a process by which the G+C content of DNA increases due to gene conversion during recombination [58].
The completeness of the gene space was quantified using BUSCO which provides a quantitative measure for genome and transcriptome completeness based on a core set of highly conserved plant-specific single-copy orthologs [59]. Of the 1440 plant-specific orthologs, 1387 (96.3%) were identified in the A. atlantica genome assembly as full length, while 1395 (96.9%) were identified in the A. eriantha assembly as full length, suggesting high-quality and complete genome assemblies. As expected for diploid species, the level of gene duplication, as identified by BUSCO for the conserved orthologous genes was low for both species (2.2% and 2.3%). Similarly, a BUSCO analysis of the transcript and protein annotation sets produced by MAKER identified similar numbers of conserved orthologs for both species, which is indicative of a successful annotation process.
Genomic features
Pericentromeric regions, associated with reduced recombination relative to physical distance, were evident from the linkage and physical map comparison in A. atlantica (Fig. 1). The observation that gene density is substantially reduced provided further evidence that that these regions represented centromeric regions in both species, as has been well-documented previously in other eukaryotes [60, 61]. Centromeres in most plant species are complex but are dominated by megabase-sized arrays of tandemly arranged monomeric satellite repeats. While complex and highly diverse among plant species, they commonly share a unit length ranging between 150 and 180 bp, which is close to the size of the nucleosome unit [62]. Melters et al. [63] showed that due to the relative size of the centromere, the most common repeat found in whole-genome sequencing data is the putative centromeric repeat. Using the output of RepeatModeler from A. eriantha, we identified a high-copy-number 159 bp tandem repeat that aligned specifically with the putative centromere location in each of the A. atlantica and A. eriantha chromosomes (Fig. 2; Additional file 2). Although the 159 bp repeat is similar in size to the putative centromeric repeats found in other grass species (e.g., B. distachyon, 156 bp; H. vulgare, 139 bp; Oryza brachyantha A.Chev. & Roehr., 154 bp; Z. mays, 156 bp), not surprisingly it shares little sequence similarity with the centromeric repeats of those species. Indeed, centromeric repeats exhibit little to no evidence of sequence similarity beyond ~ 50 million years of divergence [63]. As has been documented in other plant species, these putative centromeric repeats span a large portion (often > 50 Mb) of the A. atlantica and A. eriantha chromosomes, suggesting the presence of large pericentromeric heterochromatic regions [64, 65]. Moreover, the positioning of the centromeres, as defined by this putative repeat and the gene density plots, is consistent with the cytological positioning of the centromere, which suggests that the centromeres in A. atlantica are almost all metacentric to submetacentric, while the centromeres in A. eriantha are almost all sub-telocentric [66, 67]. Indeed, per our analyses, we identified three metacentric, two submetacentric and two sub-telocentric chromosomes in A. atlantica, and five sub-telocentric, one submetacentric and one metacentric chromosome in A. eriantha (Fig. 2).
Repeatmodeler annotated a putative telomeric satellite sequence (665 bp) for A. eriantha (Additional file 1: Table S1). A homologous sequence (639 bp) with significant homology (E-value = 0.0) and alignment identity (Identity = 80%; Gap = 6%) was identified from the repeat sequences identified by RepeatModeler (Additional file 2). BLAST searches of the assemblies with their respective satellite telomeric repeat sequence identified enriched regions of the telomeric repeat on all seven of the chromosomes for each species (Fig. 2). In A. atlantica, telomeric satellite sequences were located toward the distal end of each chromosome; however, in A. eriantha the location of the sequence is more dispersed, being found primarily at the end of the chromosomes on ten of the 14 chromosome arms, while in a few instances being interspersed interstitially. Interstitial telomere-like repeats have been reported in several plant species including Anthurium, Vicia, Sideritis, Typhonium, and Pinus where they were implicated in chromosome rearrangements, including inversions, translocations, and chromosome fusions [68,69,70,71]. While chromosomal rearrangements are common in Avena, we caution that the very repetitive nature of telomeric sequences makes them susceptible to collapse during the assembly process and are thus inherently difficult to order and orient in the Hi-C scaffolding process.
SNP discovery and genetic diversity
To characterize the diversity and phylogenetic relationships among Avena A- and C-genome diploid species, we resequenced at 10X coverage 61 A-genome diploid accessions (including A. atlantica, A. brevis, A. canariensis, A. damascena, A. hirtula, A. longiglumis, A. lusitanica, A. strigosa, A. strigosa-brevis, A. strigosa-hispanica, A. strigosa-nuda, and A. wiestii) and 10 C-genome diploids (A. clauda, A. eriantha, A. ventricosa; Additional file 3: Table S2). The resequencing produced 40 Gb sequence data per accession (Additional file 3: Table S2). The A-genome accession reads were mapped against the A. atlantica genome, while the C-genome species were mapped against the A. eriantha genome for SNP discovery using InterSnp [72]. InterSnp uses BAM files to call SNPs between samples based on consensus alleles at each genomic position, filtered to produce a dataset with 0% missing data across all lines. Considering the cleistogamous nature of the accessions included, any SNPs with > 5% heterozygous calls were deemed likely to result from spurious read-mapping and were removed from the dataset. Using a minimum allele frequency threshold of < 0.1, a total of 286,567 and 3,185,959 putative SNPs were identified within the A-genome and C-genome diploid datasets, respectively, and used by SNPhylo [73] to investigate phylogenetic relationships. SNPhylo reduces oversampling effects at linked SNPs using an LD threshold (0.1) with a sliding window of 500,000 base pairs. Thus, a total of 7221 and 11,530 SNPs, with an average of 1032 and 1647 SNPs per chromosome, were selected prior to tree construction for the A-genome and C-genome diploids dataset, respectively (Additional file 4: Table S3).
The bootstrapped maximum likelihood phylogenetic trees were rooted with either the A. eriantha accession CN 19328 for the A-genome accessions tree (Fig. 5a) or with the A. atlantica accession Cc 7277 for the C-genome accessions tree (Fig. 5b). The A-genome diploids formed two distinct clades: one of these consisted primarily of accessions classified in taxa having the AsAs subgenome, which had previously been described by Rajhathy and Morrison [74] and Leggett [75] as including A. atlantica, A. hirtula, the domesticated forms of A. strigosa, and A. wiestii; and a second clade comprised mostly of A. canariensis (AcAc), Syrian accessions of A. damascena (AdAd), A. longiglumis (AlAl), and three floret-shattering accessions that were possibly misidentified as A. hirtula and A. lusitanica. As expected, the spikelet-shattering A. atlantica occupied the basal position on the AsAs branch of the tree, while all of the A. strigosa (domesticated AsAs) genotypes formed a clade at the top of the tree and included a single accession of weedy A. wiestii (CIav 1994) that, upon inspection of the panicles, more closely resembled a long-awned, semi-shattering A. strigosa genotype.
The A. strigosa branch shows clearly the effect of a domestication bottleneck. This branch of the tree is subdivided into two distinct sub-branches. The upper sub-branch consists predominantly of genotypes of Iberian origin (i.e., CN 25698, CIav 9019, CIav 9036, etc.) and includes seven homogeneous accessions that are derivatives of the Brazilian ‘Saia’ variety of forage oat (i.e., CIav 7010, PI 291990, etc.). Interestingly, the A. hispanica genotypes form a unitary subclade within this branch that is strongly supported by the bootstrap value. The lower sub-branch is comprised of accessions from outside the Iberian Peninsula (PI 83721, PI 287314, PI 304557, CIav 9022, etc.) and includes all of the A. strigosa-nuda varieties. Since A. brevis strains are distributed in both branches, there is no evidence to confirm its identity as a distinct taxon within or apart from A. strigosa. The presence of branches containing multiple, genetically indistinct accessions indicates there is a high degree of duplication being curated within the USDA and PGR-Canada gene banks for A. strigosa.
The remainder of the A-genome tree consists of entirely wild genotypes. Avena lusitanica is not a universally accepted taxon, and the presence of these strains on various branches of the tree confirms that this is not a valid independent taxonomic entity; instead, it is part of the floret-dispersing A. hirtula-wiestii complex of semi-desert and Mediterranean scrub ecotypes of the AsAs biological species complex. The presence of three floret-shattering accessions from Morocco that were previously identified as A. damascena (AdAd) on the AsAs branch (PI 657468, PI 657471, PI 657472) and the two Syrian A. damascena genotypes on the other branch (CN 19457, CN 19459) suggests that the Moroccan group are misidentified and are therefore, like A. lusitanica, either members of the AsAs A. hirtula-wiestii-atlantica-strigosa complex or, possibly, misclassified accessions of tetraploid A. barbata.
The rooted C-genome tree had the lone A. ventricosa (CvCv) accession at the base of the C-genome branch that consisted of two subclades. The more basal branch consisted of accessions of spikelet-shattering A. eriantha (CpCp) from Algeria (CN 24022) and four samples of A. eriantha from an extended population growing in the Middle Atlas Mountains of Morocco (PI 657575–8). The other branch included Algerian (CN 24040) and Turkish (CN 19238) accessions of floret-shattering A. clauda (CpCp) along with Iranian (CN 19256) and Algerian (CN 19328, the reference genome) A. eriantha genotypes.
Discussion
Comparative genomics
The age of the Poaceae family has been difficult to establish, with varying ages reported in the literature [76, 77]. Schubert et al. [78] recently reported the use of newly available paleobotanical fossils to established the age of the family to be approximately 120.8 million years ago (Ma), with the split of the Aveneae, Brachypodieae, and BOP clades occurring approximately 44.3, 51.8, and 80.2 Ma, respectively - suggesting that the grasses have a lower nucleotide substitution rate than the other angiosperms [79]. We calculated the rate of synonymous nucleotide substitutions per synonymous site (Ks) for orthologous gene pairs between the A. atlantica and A. eriantha assemblies using the CodeML [80] tool on the CoGe platform (genomevolution.org/coge). A total of 18,002 duplicate gene pairs were identified with a clear peak seen at Ks = 0.0875 (Additional file 5: Figure S1). From the node estimates reported by Schubert et al., we calculated an average substitution rate for the Pooideae lineage of 3.39E-09, suggesting that speciation between A. atlantica and A. eriantha occurred between 5.4–12.9 million years ago (Ma), depending on whether a core eukaryotic-based synonymous mutation rate or the calculated lineage specific rate for Pooideae was used in the calculation [81, 82]. As seen in the SynMap dotplot alignment (Additional file 6: Figure S2), significant synteny was observed between the Avena chromosomes consisting of 187 syntenic blocks with 21,021 collinear genes pairs (112 genes/block) with 98.2% coverage across both the A. atlantica and A. eriantha genomes. As expected, given the relatively close ancestry of the species, the size (bp) of the syntenic blocks between species was highly correlated (R2 = 0.88; Additional file 6: Figure S2C). The large blocks of syntenic genes are suggestive of orthologous relationships between the chromosomes of the species (Additional file 6: Figure S2A). For example, slightly more than 77% (349 Mb) of the syntenic sequence found on AA2 is derived from AE5, suggesting that they are orthologs. Indeed, using a majority rule (> 50% syntenic sequence) we identified the following orthologous chromosome pairs: AA1 + AE6 (61%; 248 Mb); AA2 + AE5 (77%; 349 Mb); AA3 + AE3 (74%; 318 Mb); AA4 + AA1 (71%; 271 Mb); AA7 + AE2 (57%; 274 Mb); with AA5 and AA6 sharing orthology with several A. eriantha chromosomes (Additional file 6: Figure S2B).
The Poaceae family consists of many agronomically important species, commonly referred to as cereals, that are found in three subfamilies: Oryzoideae (rice), Panicoideae (maize, sorghum) and Pooideae (wheat, barley, oat and rye). Pooideae forms 14 tribes, including the tribes Brachypodieae, Poeae (syn Aveneae, including oat) and Triticeae (barley, rye, and wheat), with Poeae and Triticeae tribes having separated ~ 49 Ma [78]. This agrees well with the Ks analyses presented here for A. atlantica and A. eriantha and with the published Hordeum vulgare genome [83], which both show a clear peak at 0.3 – suggestive of a divergence time of 44 Ma (per the calculated lineage specific rate for Pooideae). As expected, the Ks analyses from the Avena comparisons with the B. distachyon genome (International Brachypodium Initiative, 2010) suggested a more distant divergence of 47–51 Ma for the split of the Avena–Brachypodium lineages (Additional file 5: Figure S1).
SynMap was also used to investigate syntenic relationships between the Avena and Hordeum chromosomes (Additional file 7: Figure S3 and Additional file 8: Figure S4). Although more syntenic blocks (719 and 714) were identified in the Avena–Hordeum comparisons, they were smaller – consisting of ~ 8.5 genes/block, accompanied by a lower syntenic block size correlation (R2 = 0.35 and 0.41; Additional file 7: Figure S3C and Additional file 8: Figure S4C). The decrease in block size and correlation is reflective of the more distant evolutionary relationship between the species. Nonetheless, the shared ancestry between the two Pooideae species was evident as seen by substantial synteny observed across all seven Avena–Hordeum chromosomes comparisons (Additional file 7: Figure S3A and Additional file 8: Figure S4A). As expected, large, proximal, non-syntenic regions were observed in regions corresponding to putative centromeres where gene density is substantially reduced [60, 61]. The synteny observed among the Avena and Hordeum chromosomes suggests several homeologous relationships. For example, Hordeum chromosome 1H is clearly orthologous with Avena chromosome AA2 and AE5. Indeed, of the syntenic sequence on 1H, 99% (116 Mb) was syntenic to AA2 and 85% (105 Mb) syntenic to AE5 – which is not surprising since we previously showed that AA2 and AE5 are orthologs (see above). Using a simple majority rule (> 50% syntenic sequence) the following are putative Hordeum–Avena orthologs: 1H + AA2/AE5; 2H + AA5/AE4; 3H + AA3/AE3; 6H + AA7/AE2; and 7H + AA1/AE6. The specific A. atlantica orthologs of 4H and 5H are likely AA6 and AA4, respectively; however, rearrangements obscure the likely orthologs for A. eriantha (Additional file 7: Figure S3B and Additional file 8: Figure S4B).
Bekele et al. [29] recently published a high-density, tag-level haplotype linkage map of hexaploid oat (A. sativa). This consensus linkage map increased the marker density of the former consensus map [28] consisting of 21 well-formed linkage groups, putatively corresponding to each of the 21 hexaploid oat chromosomes. To identify the ancestral subgenome groups (A-, C- and D-) for each of the 21 linkages groups we mapped the haplotag markers to both the A. atlantica and A. eriantha genomes. To avoid false hits, which is particularly problematic due to the highly repetitive nature of the oat genomes, only BLAST hits with perfect identity across the entirety of the marker sequence (e.g., zero gap openings and mismatches) were retained for downstream analyses. In total, 2119 and 969 tag-level haplotypes were mapped to the A. atlantica and A. eriantha genomes, respectively. The increased number (~2X) of tag-level haplotypes mapping successfully against the A. atlantica genome was expected since many D-subgenome haplotypes would map against the A-genome diploid, given the close phylogenetic relationship of these two subgenomes [16]. Indeed, close inspection of the mapping showed that in nearly all cases, tag-level haplotypes mapping to a specific A. atlantica chromosome were derived from two separate consensus linkage groups - presumably corresponding to homoeologs derived from A- and D-subgenomes. For example, 322 tag-level haplotypes mapped to chromosome AA1, with 153 (48%) derived from linkage group Mrg12 and 111 (35%) derived from linkage group Mrg02, which were previously identified as being derived from the A-, and D- subgenomes [16] (Table 3A). Other homoeologous chromosome pairs between the A- and D-subgenome included: Mrg33/Mrg08, Mrg18/Mrg01, Mrg05/Mrg04, Mrg24/Mrg06, Mrg23/Mrg11, Mrg12/Mrg02, and Mrg20/Mrg21. Similar mapping of the tag-level haplotypes against the A. eriantha genome elucidated linkage groups Mrg13, Mrg03, Mrg15, Mrg17, Mrg19, Mrg09 and Mrg 11 as being derived from C-subgenome (Table 3B). Interestingly, Mrg18, which we previously designated as an A-subgenome derived linkage group also showed substantial mapping to the C-genome chromosome AE7 – suggesting that this Mrg18 is actually derived from an A-subgenome/C- subgenome (A/C) intergenomic reciprocal translocation. This is a well-documented reciprocal translocation, first reported by Jellen et al. [84] where it was identified as 7C-17A. Other identifiable rearrangements include D- subgenome and C-subgenome (D/C) intergenomic exchanges on Mrg06/Mrg13, Mrg08/Mrg03, and Mrg19/Mrg28 (Table 3).
Utility of the genome assemblies
Given the genetic complexity of polyploid species, diploid species have frequently been used as simplified genetic models [85,86,87]. We show the value of these diploid assemblies using published genome wide association studies (GWAS) for heading date and crown rust resistance - both major breeding targets for common oat. Heading date (flowering time) is critically important for regional adaptation, photosynthetic efficiency, and stress avoidance; and through these factors it strongly influences overall yield [88]. A Haplotag-based GWAS of heading date in the CORE diversity panel (n = 635) of common hexaploid (AACCDD) oat identified two major associations on linkage groups Mrg02, at position 34 cM in eight field trails and on Mrg12 at position 40–42 cM in seven field trials [29]. Interestingly, our comparative analysis (see above) suggests that Mrg02 and Mrg12 are homoeologous (Table 3A), with Mrg12 and Mrg02 being derived from the A-subgenome and D-subgenome, respectively. BLAST searches against the A. atlantica genome assembly using the maker sequences associated with heading date on Mrg12 localized the heading date quantitative trail loci (QTL) to chromosome AA1, at an interval spanning bases 548,905,448 – 553,755,648. A total of 175 annotated gene sequence are found within this region, including a likely candidate gene at the center of this interval, AA006173 (Fig. 3a; 550,704,569–555,704,964), which is annotated as being homologous to HD3A (Heading Date 3A) from O. sativa, and is homologous (E-value = 9e-125, Identity = 97%) to the flowering time protein (FT-like; AAZ38709.1). Yan et al. [89] described HD3A as the vernalization gene, VRN3, in wheat and barley. Interestingly, while Mrg02 is likely of a D-subgenome origin, BLAST search of markers associated with heading date from the Mrg02 linkage mapped significantly to the A-genome chromosome AA1 at an interval spanning 550,053,072–550,947,435 bp, only 242,471 bp from the aforementioned HD3A gene, suggesting that the candidate gene for both major QTLs for flowering time are functional homoeologs of the flowering time (FT) HD3A gene in the A- and D-subgenomes (Fig. 3b).

Identification of candidate genes putatively underlying heading date in oats. Candidate gene loci were identified using BLAST searches against the A. atlantica genome assembly using maker sequences associated with heading date QTLs located on the homoeologous linkage groups a Mrg12 and b Mrg02 (Bekele et al. [29]). Markers from both QTLs mapped to the same physical position on chromosome AA1, within an interval containing an FT-like protein (HD3A), suggesting that heading date in modern oat is controlled by two functional homoeologs of the flowering time gene
Crown rust, caused by Puccinia coronata f. Sp. avenae, is the most damaging and widespread disease of oat worldwide [90]. Moderate to severe outbreaks can reduce yield by 10–40% [91]. Klos et al. [30] performed a GWAS of crown rust resistance on elite common oat accessions challenged with multiple P. coronata isolates and identified multiple QTL on 12 linkage groups that were associated with crown rust resistance, several of which were associated with known resistance genes (e.g., Pc91). Resistance gene analogs (RGAs) contain specific conserved domains and motifs that can be used to identify and classify R-genes into four main RGA families: specifically, NBS-encoding proteins, receptor-like protein kinases (RLKs), receptor-like proteins (RLPs), and transmembrane coiled-coil proteins (TMCC). The RGAugury pipeline [92] annotated a total of 1563 (511 NBS, 722 RLK; 120 RLP; 160 TMCC) and 1402 (459 NBS; 654 RLK; 135 RLP; 154 TMCC) RGAs in the A. atlantica and A. eriantha genomes, respectively (Additional file 9: Table S4; Additional file 10: Figure S5). As has been observed in other monocots, no Toll/Interleukin-1 receptor-NBS-LRR R-genes were predicted in either genome, supporting the hypothesis that this class of R-gene evolved in eudicot lineage [93] or were lost during the evolution of the monocots [94]. The RGAs, specifically the NBS-encoding RGAs, cluster primarily in subtelomeric regions (Additional file 10: Figure S5), with clusters identified on almost all chromosomes and often correlated with the mapping position of crown rust QTLs. For example the Pc91 gene, a known seedling resistance gene previously associated with QTL QPc.CORE.18.3 [30] maps, via two SNPs (GMI_ES03_c2277_336 and GMI_ES05_c11155_383), to the A. atlantica chromosome AA2 at positions 510,519,361 and 533,475,317, co-locating with a predicted disease gene cluster (Fig. 4). The closest annotated disease resistance genes to these markers are AA013376, similar to RPH8A (a nonfunctional homolog of rpp8 in Arabidopsis [95]) located at position 510,828,316 and AA014151, similar to RPM1 (a well-documented resistance gene in Arabidopsis [96]) located at 533,698,614 (Additional file 11: Table S5). Both candidate RGAs were identified by the RGAugury pipeline as CC-NBS-LRR containing R-genes. We caution that while these two candidate RGAs are positioned in the immediate vicinity of the associated markers, at least 87 RGA are present at the RGA cluster defined by the QTL. We note that the diagnostic SCAR and DART markers developed for Pc91 also map to this same location (527,126,948 [97, 98];).

Identification of candidate genes putatively underlying crown rust resistance in oats. Candidate gene loci were identified using BLAST searches against the A. atlantica genome assembly using maker sequences associated with crown rust QTLs located on hexaploid A. sativa linkage group Mrg18 reported by Klos et al. [30]. Mrg18 was previously shown to be involved with an intergenomic translocation involving 7C and 17A, corresponding to A. eriantha chromosome AE7 (blue) and A. atlantica chromosome AA2 (red)
Conclusions
Reference-quality, de novo whole-genome sequence assemblies for two highly repetitive ~ 4 Gb Avena diploid species were produced using a hybrid approach involving PacBio long reads, Illumina short reads, and both in vitro and in vivo chromatin-contact mapping. The whole-genome reference assemblies for As- and Cp-genome oat diploids provided for the first time in this paper represent powerful tools for identifying genes that underlie adaptive, disease resistance, and grain-quality traits critical for oat improvement. The utility of these whole-genome references was demonstrated first by analyzing sequences homologous to heading-date QTL-containing regions that were previously identified via GWAS in common hexaploid oat (A. sativa) to find linked candidate genes in A. atlantica and A. eriantha. Additionally, we used these references in successfully identifying RGAs homologous to oat crown rust resistance genes.
Avena atlantica retains a remarkable degree of synteny in comparison with barley while A. eriantha has undergone a relatively greater degree of chromosomal rearrangement, suggesting the presence of an underlying genomic instability in the C-genome diploids. This might be related to the abundant heterochromatin, including the underlying pAm1 repeat motif, distributed throughout the chromosomes of this genome (Fig. 2b, Track 5 [27];). Their genome sequences shed enormous insight into the complex evolutionary processes that have led to the appearance of cultivated diploid, tetraploid, and hexaploid oat going back millions of years. These processes included responses to natural selective events such as the Zanclean Cataclysm ~ 5 Ma and repeated cycles of global climate change characterized by boreal glacial maxima interspersed with humid periods and desertification due to northerly expansions of the Saharan and Arabian Deserts [99,100,101].
We demonstrate that A. atlantica, A. strigosa, and A. wiestii represent multiple ecotypes or subspecies of a single biological species complex sharing the subgenome designation AsAs and distinguished primarily by their seed-dispersal strategies. The phylogeny presented here, which was generated by analyzing thousands of SNPs identified via resequencing of dozens of geographically diverse accessions, clearly illustrates a monophyletic relationship with A. atlantica accessions at the root of the AsAs-genome clade (Fig. 5a). This is further seen in the high degree of synteny and collinearity between the A. atlantica chromosomes and A. strigosa X wiestii linkage groups reported by Kremer et al. [102] (Additional file 12: Figure S6). This result is remarkable, given the high degree of chromosomal rearrangement previously observed among different species and genomes within Avena [27].

Abbreviated maximum likelihood tree generated using a 10,894 SNPs for C-genome diploids rooted to the A. atlantica (AT_Cc7277) reference and b 7221 SNPs for A-genome diploids rooted to the A. eriantha reference (ER_CN 19238). Asterisks denote percentage of 1000 bootstrap replicates that support the topology at 90–100% (gold) and 75–89% (blue). Scale bar represents substitutions per site. Branch labels are based on subgenome composition and, in some cases, diaspore morphology (“floret-shattering,” “spikelet-shattering,” or “cultivated”). Unabbreviated trees are provided as Additional file 15: Figure S7 and Additional file 16: Figure S8
The oat community has struggled without a reference genome for decades. Finally, we have complete references for what are, essentially, all three component genomes of cultivated hexaploid oat and the four known subgenomes of the genus, given the close correspondence between Avena subgenomes A, B, and D. Moreover, once a complete hexaploid reference is available, the utility of these component genomes will increase further, as they will provide a precise roadmap of the structural and functional evolutionary steps that took place in the formation of this unique and important polyploid species.
Methods
Plant material and nucleic acid extraction
For whole-genome assembly, young leaf tissue (~ 14–21 days post emergence), dark treated for 72 h, from A. atlantica (CC7277; T. Langdon, Aberystwyth University, Wales, UK) and A. eriantha (BYU132; EN Jellen, Brigham Young University, Provo, UT) was flash-frozen and sent to the Arizona Genomics Institute (AGI; Tucson, AZ, USA) for high molecular weight DNA extraction. For the diversity panel, DNA from 76 accessions of diploid A- and C-subgenome species (Additional file 3: Table S2) was extracted from 30 mg of freeze-dried leaf tissue using a protocol devised by Sambrook et al. [103] with modifications described by Todd and Vodkin [104]. All plants were grown in greenhouses at Brigham Young University (BYU) using Sunshine Mix II (Sun Gro, Bellevue, WA, USA) supplemented with Osmocote fertilizers (Scotts, Marysville, OH, USA) and maintained at 25 °C under broad-spectrum halogen lamps, with 12-h photoperiods.
DNA sequencing and read processing
For whole-genome sequencing, large-insert SMRTBell libraries (> 20 kb), selected using a BluePippin System (Sage Science, Inc., Beverly, MA, USA), were prepared according to standard manufacture protocols. Libraries were sequenced using P6-C4 chemistry on either the RS II or Sequel sequencing instruments (Pacific BioSciences, Menlo Park, CA, USA; Additional file 13: Table S6). Sequencing was performed for A. atlantica at the DNA Sequencing Center (DNASC) at BYU (Provo, UT, USA) and at RTL Genomics (Lubbock, TX), while the sequencing for A. eriantha was performed at the BYU DNASC. For the diversity panel and for whole-genome polishing, extracted DNA was sent to the Beijing Genomic Institute (BGI; Hong Kong, China) for 2 × 150 bp paired end (PE) sequencing from standard 500-bp insert libraries. Trimmomatic v0.35 [105] was used to remove adapter sequences and leading and trailing bases with a quality score below 20 or average per-base quality of 20 over a four-nucleotide sliding window. After trimming, any reads shorter than 75 nucleotides in length were removed.
RNA-Seq and transcriptome assembly
For A. atlantica, RNA-Seq data consisted 2 × 100 bp PE Illumina reads derived from 11 different plant tissue types including, stem, mature leaf, stressed mature leaf, seed (2 days old), hypocotyl (4–5 days old), root (4–5 days old), vegetative meristem, green grain, yellow grain, young flower (meiotic), and green anthers (Additional file 14: Table S7). For A. eriantha, 2 × 150 PE RNA-Seq data was generated by BGI from six tissue sources, including young leaf, mature leaf, crown, roots, immature panicle, and whole seedling, harvested from plants grown hydroponically in 1× Maxigro™ (GH Inc., Sebastopol, CA, USA) in growth chambers maintained at 21 °C under broad-spectrum halogen lamps, with a 12-h photoperiod at BYU (Additional file 14: Table S7). The resulting reads were trimmed using Trimmomatic [105], then aligned to either the A. atlantica or A. eriantha reference using HiSat2 v2.0.4 [106] with default parameters and max intron length set to 50,000 bp. Following alignment, the resulting SAM file was sorted and indexed using SAMtools v1.6 [107] and assembled into putative transcripts using StringTie v1.3.4 [108]. The quality of the assembled transcriptome was assessed relative to completeness using BLAST comparisons to the reference Brachypodium distachyon L. (ftp://ftp.ensemblgenomes.org/pub/plants/release-37/fasta/brachypodium_distachyon/pep/).
Genome size, assembly, polishing, and scaffolding
Genome size was estimated using Jellyfish [109] and GenomeScope v1.0 [31] at k-mer length = 21 for each species. Initial assemblies were done using Canu v1.7 [33] with default parameters (corMhapSensitivity = normal and corOutCoverage = 40). The resulting assemblies were polished using Arrow from the GenomicConsensus package in the Pacific BioSciences SMRT portal v5.1.0 and PILON v0.22 [110] using Illumina short reads. Chicago® and Hi-C proximity-guided assemblies were performed by Dovetail Genomics LLC (Santa Cruz, CA, USA) to produce chromosome-scale assemblies. Fresh leaf tissue from a single dark-treated (72 h), 3-week-old plant, derived directly from selfing of the original A. atlantica and A. eriantha plants, was sent to Dovetail Genomics for Chicago® and Hi-C library preparation as described by Putnam et al. [35] and Lieberman-Aiden et al. [111], respectively, using the DpnII restriction endonuclease. The libraries were sequenced using a standard Illumina library prep followed by sequencing on an Illumina HiSeq X in rapid run mode. The HiRiSE™ scaffolder and the Chicago® and Hi-C library-based read pairs were used to a produce a likelihood model for genomic distance between read pairs, which was used to break putative miss-joins and to identify and make prospective joins in the de novo Canu assemblies.
Repeat analysis, genome completeness, and annotation
RepeatModeler v1.0.11 [112] and RepeatMasker v4.0.7 [113] were used to quantify and classify repetitive elements in the final assemblies, relative to RepBase libraries v20181026; www.girinst.org). Benchmarking Universal Single-Copy Orthologs (BUSCO) v3.0.2 [59] was employed to assess the completeness of the assembly using the Embryophyta odb9 dataset and the “—long” argument, which applies Augustus [114] optimization for self-training.
MAKER2 v2.31.10 [56, 57] was used to annotate the final genomes. Expressed sequence tag evidence for annotation included the de novo transcriptomes for each species as well as the cDNA models from Brachypodium distachyon L. (v 1.0; Ensembl genomes). Protein evidence included the uniprot-sprot database (downloaded September 25, 2018) as well as the peptide models from B. distachyon (v 1.0; Ensembl genomes). Repeats were masked based on species-specific files produced by RepeatModeler. For ab initio gene prediction, A. atlantica and A. eriantha species-specific AUGUSTUS gene prediction models were provided as well as rice (Oryza sativa)-based SNAP models.
Variant identification and tree creation
Single nucleotide polymorphisms (SNPs) for the diversity panel were identified from the Illumina reads by mapping the A-subgenome and C-subgenome diploid accessions against the A. atlantica and A. eriantha reference genome assemblies, respectively, using BWA-mem v0.7.17 [115]. Output SAM files were converted to BAM files and sorted using SAMtools v1.6 [107], and indexed using Sambamba v0.6.8 [116]. InterSnp, an analysis tool from the BamBam v1.4 package [72], was used to call SNPs with the arguments -m 2 and -f 0.35. Bash scripting was used to removed SNPs with less than 100% genotype calls across all accessions (i.e., no missing data) or given the cleistogamous nature of the species where 5% or more of the accessions were called as heterozygotes. SNPs on unscaffolded contigs were also removed prior to phylogenetic analysis. SNPhylo v20160204 [73], which uses MUSCLE [117] for sequence alignments and linkage disequilibrium to down sample the SNP dataset, was used to build Phylogenies with the bootstrapping parameter set to 1000. The resulting tree was visualized using FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree).
Genome comparison
Genomic comparisons, including calculations of synonymous substitutions per synonymous sites (Ks) and homology searches for syntenic gene-sets with Hordeum vulgare L. (CoGe genome id52970 [118]), Oryza sativa L. (CoGe genome id34910 [119]), Zea mays L. (CoGe genome id33766 [120]), and B. distachyon (CoGe genome id52735 Vogel [121]) were accomplished using the DAGchainer output file from the CoGe (https://genomevolution.org/coge/) SynMap tool.
Availability of data and materials
All data generated or analyzed during this study are included in this published article, its supplementary information files and publicly available repositories. The raw sequences used for A. atlantica genome assembly are deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive database under the BioProject PRJNA546592 [122] with the following accession numbers: SRR9720684 (PacBio reads), SRR9841448–SRR9841455 (Hi-C reads), SRR9841587–SRR9841597 (Transcriptome). Similarly, the raw sequences for the A. eriantha genome assembly are found in BioProject PRJNA546595 [123] with the following accession numbers: SRR9720373 (PacBio reads), SRR9833273–SRR9833276 (Hi-C reads), SRR9722223 (Polishing short reads), SRR9722219–SRR9722222, SRR9722225, SRR9722226 (Transcriptome). The raw reads for the resequencing panel of the diploid species (Additional file 3: Table S2) are found in BioProject PRJNA556219 [124] with the following NCBI accession numbers: SRR9933122–SRR9933198 (resequencing panel). Genome browsing and bulk data downloads, including annotations and BLAST analysis of the final proximity-guided assemblies are available at CoGe (https://genomevolution.org/coge/) with genome IDs: id53337 (A. atlantica [125]) and id53381 (A. eriantha [126]).
Abbreviations
- BLAST:
-
Basic Local Alignment Search Tool
- GWAS:
-
Genome-wide association studies
- LG:
-
Linkage group
- Ma:
-
Million years ago
- QTL:
-
Quantitative trail loci
- RGA:
-
Resistance gene analog
- SNPs:
-
Single nucleotide polymorphisms
References
Ahmad M, Zaffar G, Mir SD, Razvi SM, Dar ZA, Iqbal S. Genetic analysis for fodder yield and other important traits in oats (Avena sativa L.). Indian J Genet Pl Br. 2014;74(1):112–4.
Oliver RE, Tinker NA, Lazo GR, Chao S, Jellen EN, Carson ML, Rines HW, Obert DE, Lutz JD, Shackelford I, et al. SNP discovery and chromosome anchoring provide the first physically-anchored hexaploid oat map and reveal synteny with model species. PLoS One. 2013;8(3):e58068.
Andon MB, Anderson JW. State of the art reviews: the oatmeal-cholesterol connection: 10 years later. Am J Lifestyle Med. 2008;2(1):51–7.
Jenkins AL, Jenkins DJA, Zdravkovic U, Wursch P, Vuksan V. Depression of the glycemic index by high levels of beta-glucan fiber in two functional foods tested in type 2 diabetes. Eur J Clin Nutr. 2002;56(7):622–8.
Yarnell E, Abascal K. Botanical remedies for nicotine addiction. Altern Complement Ther. 2001;7(6):337–40.
Yarnell EaA K. Botanical treatments for depression. Altern Complement Ther. 2001;7(3):138–43.
Fardet A. New hypotheses for the health-protective mechanisms of whole-grain cereals: what is beyond fibre? Nutr Res Rev. 2010;23(1):65–134.
Peterson DM. Oat antioxidants. J Cereal Sci. 2001;33(2):115–29.
Daou C, Zhang H. Oat Beta-Glucan: its role in health promotion and prevention of diseases. Compr Rev Food Sci F. 2012;11(4):355–65.
Grimalt R, Mengeaud V, Cambazard F, Study Investigators G. The steroid-sparing effect of an emollient therapy in infants with atopic dermatitis: a randomized controlled study. Dermatology. 2007;214(1):61–7.
Potter RC, Castro JM, L.C. M: Oat oil compositions with useful cosmetic and dermatological properties in. Edited by States U, vol. US5620692A. United States: GTC OATS Inc 1997.
Singh R, De S, Belkheir A. Avena sativa (oat), a potential Neutraceutical and therapeutic agent: an overview. Crit Rev Food Sci. 2013;53(2):126–44.
Sur R, Nigam A, Grote D, Liebel F, Southall MD. Avenanthramides, polyphenols from oats, exhibit anti-inflammatory and anti-itch activity. Arch Dermatol Res. 2008;300(10):569–74.
Soreng RJ, Peterson PM, Romaschenko K, Davidse G, Teisher JK, Clark LG, Barbera P, Gillespie LJ, Zuloaga FO. A worldwide phylogenetic classification of the Poaceae (Gramineae) II: an update and a comparison of two 2015 classifications. J Syst Evol. 2017;55(4):259–90.
Zhou X, Jellen EN, Murphy JP. Progenitor germplasm of domesticated hexaploid oat. Crop Sci. 1999;39(4):1208–14.
Yan H, Bekele WA, Wight CP, Peng Y, Langdon T, Latta RG, Fu YB, Diederichsen A, Howarth CJ, Jellen EN, et al. High-density marker profiling confirms ancestral genomes of Avena species and identifies D-genome chromosomes of hexaploid oat. Theor Appl Genet. 2016;129(11):2133–49.
Loskutov IG, Rines HW. Avena. In: Kole C, editor. Wild Crop Relatives: Genomic and Breeding Resources. Heidelberg: Springer; 2011. p. 109–83.
Aung T, Chong J, Leggett M. The transfer of crown rust resistance Pc94 from a wild diploid to cultivated hexaploid oat. In: Kema GHJ, Niks RE, Daamen RA (eds) Proc. 9th Int. Eur. Mediterr. Cereal Rusts and Powdery Mildews Conf. Lunteren Netherlands. Wageningen, European and Mediterranean Cereal Rust Foundation. 1996. pp. 167–71.
Dyck PL, Zillinsky FJ. Inheritance of Crown Rust Resistance Transferred from Diploid to Hexaploid Oats. Can J Genet Cytol. 1963;5(5):398.
Welch RW, Brown JCW, Leggett JM. Interspecific and intraspecific variation in grain and great characteristics of wild oat (Avena) species: Very high great (1 -> 3),(1 -> 4)-beta-D-glucan in an Avena atlantica genotype. J Cereal Sci. 2000;31(3):273–9.
Jellen EN, Beard J. Geographical distribution of a chromosome 7C and 17 intergenomic translocation in cultivated oat. Crop Sci. 2000;40(1):256–63.
Fominaya A, Vega C, Ferrer E. Giemsa C-banded karyotypes of Avena species. Genome. 1988;30(5):627–32.
Coon MA. Characterization and variable expression of the CslF6 homologs in oat (Avena sp.). Provo: Brigham Young University; 2012.
Jellen EN, Gill BS, Cox TS. Genomic in-situ hybridization differentiates between a/D-genome and C-genome chromatin and detects Intergenomic translocations in Polyploid oat species (genus Avena). Genome. 1994;37(4):613–8.
Oliver RE, Jellen EN, Ladizinsky G, Korol AB, Kilian A, Beard JL, Dumlupinar Z, Wisniewski-Morehead NH, Svedin E, Coon M, et al. New diversity arrays technology (DArT) markers for tetraploid oat (Avena magna Murphy et Terrell) provide the first complete oat linkage map and markers linked to domestication genes from hexaploid a. sativa L. Theor Appl Genet. 2011;123(7):1159–71.
Bennett MD, Smith JB. Nuclear dna amounts in angiosperms. Philos Trans R Soc Lond Ser B Biol Sci. 1976;274(933):227–74.
Sanz MJ, Jellen EN, Loarce Y, Irigoyen ML, Ferrer E, Fominaya A. A new chromosome nomenclature system for oat (Avena sativa L. and A. byzantina C. Koch) based on FISH analysis of monosomic lines. Theor Appl Genet. 2010;121(8):1541–52.
Chaffin AS, Huang YF, Smith S, Bekele WA, Babiker E, Gnanesh BN, Foresman BJ, Blanchard SG, Jay JJ, Reid RW et al. A Consensus Map in Cultivated Hexaploid Oat Reveals Conserved Grass Synteny with Substantial Subgenome Rearrangement. Plant Genome. 2016;9(2):1–21.
Bekele WA, Wight CP, Chao SM, Howarth CJ, Tinker NA. Haplotype-based genotyping-by-sequencing in oat genome research. Plant Biotechnol J. 2018;16(8):1452–63.
Klos KE, Yimer BA, Babiker EM, Beattie AD, Bonman JM, Carson ML, Chong J, Harrison SA, Ibrahim AMH, Kolb FL et al. Genome-Wide Association Mapping of Crown Rust Resistance in Oat Elite Germplasm. Plant Genome. 2017;10(2):1–13.
Vurture GW, Sedlazeck FJ, Nattestad M, Underwood CJ, Fang H, Gurtowski J, Schatz MC. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 2017;33(14):2202–4.
Yan H, Martin SL, Bekele WA, Latta RG, Diederichsen A, Peng Y, Tinker NA. Genome size variation in the genus Avena. Genome. 2016;59(3):209–20.
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–36.
Singh R, Ming R, Yu QY. Comparative analysis of GC content variations in plant genomes. Trop Plant Biol. 2016;9(3):136–49.
Putnam NH, O'Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, Troll CJ, Fields A, Hartley PD, Sugnet CW, et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016;26(3):342–50.
Latta RG, Bekele WA, Wight CP, Tinker NA: Comparative linkage mapping of diploid, tetraploid, and hexaploid Avena species suggests extensive chromosome rearrangement in ancestral diploids. Sci Rep 2019, In Press.
Tinker NA, Bekele WA, Hattori J. Haplotag: Software for Haplotype-Based Genotyping-by-Sequencing Analysis. G3-Genes Genom Genet. 2016;6(4):857–63.
Haupt W, Fischer TC, Winderl S, Fransz P, Torres-Ruiz RA. The centromere1 (CEN1) region of Arabidopsis thaliana: architecture and functional impact of chromatin. Plant J. 2001;27(4):285–96.
Talbert PB, Henikoff S. Centromeres convert but don't cross. PLoS Biol. 2010;8(3):e1000326.
Du J, Tian Z, Hans CS, Laten HM, Cannon SB, Jackson SA, Shoemaker RC, Ma J. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. Plant J. 2010;63(4):584–98.
Galindo-Gonzalez L, Mhiri C, Deyholos MK, Grandbastien MA. LTR-retrotransposons in plants: engines of evolution. Gene. 2017;626:14–25.
Tenaillon MI, Hollister JD, Gaut BS. A triptych of the evolution of plant transposable elements. Trends Plant Sci. 2010;15(8):471–8.
Ibarra-Laclette E, Lyons E, Hernandez-Guzman G, Perez-Torres CA, Carretero-Paulet L, Chang TH, Lan T, Welch AJ, Juarez MJ, Simpson J, et al. Architecture and evolution of a minute plant genome. Nature. 2013;498(7452):94–8.
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326(5956):1112–5.
Tian ZX, Rizzon C, Du JC, Zhu LC, Bennetzen JL, Jackson SA, Gaut BS, Ma JX. Do genetic recombination and gene density shape the pattern of DNA elimination in rice long terminal repeat retrotransposons? Genome Res. 2009;19(12):2221–30.
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457(7229):551–6.
Baucom RS, Estill JC, Chaparro C, Upshaw N, Jogi A, Deragon JM, Westerman RP, Sanmiguel PJ, Bennetzen JL. Exceptional diversity, non-random distribution, and rapid evolution of retroelements in the B73 maize genome. PLoS Genet. 2009;5(11):e1000732.
Langdon T, Jenkins G, Hasterok R, Jones RN, King LP. A high-copy-number CACTA family transposon in temperate grasses and cereals. Genetics. 2003;163(3):1097–108.
Wicker T, Guyot R, Yahiaoui N, Keller B. CACTA transposons in Triticeae. A diverse family of high-copy repetitive elements. Plant Physiol. 2003;132(1):52–63.
Bilinski P, Han Y, Hufford MB, Lorant A, Zhang P, Estep MC, Jiang J, Ross-Ibarra J. Genomic abundance is not predictive of tandem repeat localization in grass genomes. PLoS One. 2017;12(6):e0177896.
Feschotte C, Swamy L, Wessler SR. Genome-wide analysis of mariner-like transposable elements in rice reveals complex relationships with stowaway miniature inverted repeat transposable elements (MITEs). Genetics. 2003;163(2):747–58.
Minaya M, Pimentel M, Mason-Gamer R, Catalan P. Distribution and evolutionary dynamics of stowaway miniature inverted repeat transposable elements (MITEs) in grasses. Mol Phylogenet Evol. 2013;68(1):106–18.
Solano R, Hueros G, Fominaya A, Ferrer E. Organization of repeated sequences in species of the genus Avena. Theor Appl Genet. 1992;83(5):602–7.
Katsiotis A, Loukas M, Heslop-Harrison JS. Repetitive DNA, genome and species relationships in Avena and Arrhenatherum (Poaceae). Ann Bot-London. 2000;86(6):1135–42.
Michael TP. Plant genome size variation: bloating and purging DNA. Brief Funct Genomics. 2014;13(4):308–17.
Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sanchez Alvarado A, Yandell M. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–96.
Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 2011;12:491.
Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311.
Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2.
Mizuno H, Kawahara Y, Wu JZ, Katayose Y, Kanamori H, Ikawa H, Itoh T, Sasaki T, Matsumoto T. Asymmetric distribution of gene expression in the centromeric region of rice chromosome 5. Front Plant Sci. 2011;2(16)1–12.
Philippe R, Paux E, Bertin I, Sourdille P, Choulet F, Laugier C, Simkova H, Safar J, Bellec A, Vautrin S et al: A high density physical map of chromosome 1BL supports evolutionary studies, map-based cloning and sequencing in wheat. Genome Biol. 2013;14(6):1–22.
Henikoff S, Ahmad K, Malik HS. The centromere paradox: stable inheritance with rapidly evolving DNA. Science. 2001;293(5532):1098–102.
Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby JG, Sebra R, Peluso P, Eid J, Rank D et al: Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013;14(1):1–20.
Willing EM, Rawat V, Mandakova T, Maumus F, James GV, Nordstrom KJV, Becker C, Warthmann N, Chica C, Szarzynska B, et al. Genome expansion of Arabis alpina linked with retrotransposition and reduced symmetric DNA methylation. Nat Plants. 2015;1(2):1–7.
Gan X, Hay A, Kwantes M, Haberer G, Hallab A, Dello Ioio R, Hofhuis H, Pieper B, Cartolano M, Neumann U et al: The Cardamine hirsuta genome offers insight into the evolution of morphological diversity. Nat Plants. 2016;2(11):1–6.
Baum BR, Fedak G. Avena-Atlantica, a new diploid species of the oat genus from Morocco. Can J Bot. 1985;63(6):1057–60.
Rajhathy T, Dyck PL. Chromosomal Differentiation and Speciation in Diploid Avena .2. Karyotype of A. pilosa. Can J Genet Cytol. 1963;5(2):175.
Schubert I, Schrieverschwemmer G, Werner T, Adler ID. Telomeric signals in Robertsonian fusion and fission chromosomes - implications for the origin of Pseudoaneuploidy. Cytogenet Cell Genet. 1992;59(1):6–9.
Islam-Faridi MN, Nelson CD, Kubisiak TL. Reference karyotype and cytomolecular map for loblolly pine (Pinus taeda L.). Genome. 2007;50(2):241–51.
Raskina O, Barber JC, Nevo E, Belyayev A. Repetitive DNA and chromosomal rearrangements: speciation-related events in plant genomes. Cytogenet Genome Res. 2008;120(3–4):351–7.
Sousa A, Cusimano N, Renner SS. Combining FISH and model-based predictions to understand chromosome evolution in Typhonium (Araceae). Ann Bot. 2014;113(4):669–80.
Page JT, Liechty ZS, Huynh MD, Udall JA. BamBam: genome sequence analysis tools for biologists. BMC Res Notes. 2014;7:829.
Lee TH, Guo H, Wang X, Kim C, Paterson AH. SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics. 2014;15:162.
Rajhathy T, Morrison JW. Chromosome morphology in the genus Avena. Can J Bot. 1959;37(3):331–7.
Leggett JM. Interspecific hybrids involving the recently described diploid taxon Avena-Atlantica. Genome. 1987;29(2):361–4.
Bremer K. Ancestral areas - a Cladistic reinterpretation of the Center of Origin Concept. Syst Biol. 1992;41(4):436–45.
Bouchenak-Khelladi Y, Verboom GA, Savolainen V, Hodkinson TR. Biogeography of the grasses (Poaceae): a phylogenetic approach to reveal evolutionary history in geographical space and geological time. Bot J Linn Soc. 2010;162(4):543–57.
Schubert M, Marcussen T, Meseguer AS, Fjellheim S. The grass subfamily Pooideae: cretaceous-Palaeocene origin and climate-driven Cenozoic diversification. Glob Ecol Biogeogr. 2019;28(8):1168–82.
Christin PA, Spriggs E, Osborne CP, Stromberg CAE, Salamin N, Edwards EJ. Molecular dating, evolutionary rates, and the age of the grasses. Syst Biol. 2014;63(2):153–65.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91.
Koch MA, Haubold B, Mitchell-Olds T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol Biol Evol. 2000;17(10):1483–98.
Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–5.
Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T, Radchuk V, Dockter C, Hedley PE, Russell J, et al. A chromosome conformation capture ordered sequence of the barley genome. Nature. 2017;544(7651):426.
Jellen EN, Gill BS, Rines HW, Fox SL, Wilson WA, McMullen MS. Translocations in current and ancestral spring and winter oat accessions. In: 1996 Agronomy abstracts, vol. 1996. Madison: Agronomy Society of America. p. 78.
Jarvis DE, Ho YS, Lightfoot DJ, Schmockel SM, Li B, Borm TJA, Ohyanagi H, Mineta K, Michell CT, Saber N, et al. The genome of Chenopodium quinoa. Nature. 2017;542(7641):307.
Du XM, Huang G, He SP, Yang ZE, Sun GF, Ma XF, Li N, Zhang XY, Sun JL, Liu M, et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat Genet. 2018;50(6):796.
Bertioli DJ, Cannon SB, Froenicke L, Huang GD, Farmer AD, Cannon EKS, Liu X, Gao DY, Clevenger J, Dash S, et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet. 2016;48(4):438.
Mathan J, Bhattacharya J, Ranjan A. Enhancing crop yield by optimizing plant developmental features. Development. 2016;143(18):3283–94.
Yan L, Fu D, Li C, Blechl A, Tranquilli G, Bonafede M, Sanchez A, Valarik M, Yasuda S, Dubcovsky J. The wheat and barley vernalization gene VRN3 is an orthologue of FT. P Natl Acad Sci USA. 2006;103(51):19581–6.
Carson ML. Virulence in oat crown rust (Puccinia coronata f. sp avenae) in the United States from 2006 through 2009. Plant Dis. 2011;95(12):1528–34.
Simmons MD. The Cereal Rusts Vol II: Diseases, distribution, epidemiology and control. Orlando: Academic Press; 1985.
Li PC, Quan XD, Jia GF, Xiao J, Cloutier S, You FM. RGAugury: a pipeline for genome-wide prediction of resistance gene analogs (RGAs) in plants. BMC Genomics. 2016;17(852):1–10.
Akita M, Valkonen JPT. A novel gene family in moss (Physcomitrella patens) shows sequence homology and a phylogenetic relationship with the TIR-NBS class of plant disease resistance genes. J Mol Evol. 2002;55(5):595–605.
Bai J, Pennill LA, Ning J, Lee SW, Ramalingam J, Webb CA, Zhao B, Sun Q, Nelson JC, Leach JE, et al. Diversity in nucleotide binding site-leucine-rich repeat genes in cereals. Genome Res. 2002;12(12):1871–84.
McDowell JM, Dhandaydham M, Long TA, Aarts MGM, Goff S, Holub EB, Dangl JL. Intragenic recombination and diversifying selection contribute to the evolution of downy mildew resistance at the RPP8 locus of arabidopsis. Plant Cell. 1998;10(11):1861–74.
Grant MR, Godiard L, Straube E, Ashfield T, Lewald J, Sattler A, Innes RW, Dangl JL. Structure of the Arabidopsis Rpm1 gene enabling dual-specificity disease resistance. Science. 1995;269(5225):843–6.
McCartney CA, Stonehouse RG, Rossnagel BG, Eckstein PE, Scoles GJ, Zatorski T, Beattie AD, Chong J. Mapping of the oat crown rust resistance gene Pc91. Theor Appl Genet. 2011;122(2):317–25.
Gnanesh BN, Fetch JM, Menzies JG, Beattie AD, Eckstein PE, McCartney CA. Chromosome location and allele-specific PCR markers for marker-assisted selection of the oat crown rust resistance gene Pc91. Mol Breeding. 2013;32(3):679–86.
Garcia-Castellanos D, Estrada F, Jimenez-Munt I, Gorini C, Fernandez M, Verges J, De Vicente R. Catastrophic flood of the Mediterranean after the Messinian salinity crisis. Nature. 2009;462(7274):778–U796.
Blanc PL. The opening of the Plio-quaternary Gibraltar Strait: assessing the size of a cataclysm. Geodin Acta. 2002;15(5–6):303–17.
Kropelin S, Verschuren D, Lezine AM, Eggermont H, Cocquyt C, Francus P, Cazet JP, Fagot M, Rumes B, Russell JM, et al. Climate-driven ecosystem succession in the Sahara: the past 6000 years. Science. 2008;320(5877):765–8.
Kremer CA, Lee M, Holland JB. A restriction fragment length polymorphism based linkage map of a diploid Avena recombinant inbred line population. Genome. 2001;44(2):192–204.
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1989.
Todd JJ, Vodkin LO. Duplications that suppress and deletions that restore expression from a Chalcone synthase multigene family. Plant Cell. 1996;8(4):687–99.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome project data processing S. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650–67.
Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27(6):764–70.
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–93.
Smit A, Hubley, R: RepeatModeler Open-1.0. 2008-2015, <http://www.repeatmasker.org>. Accessed 22 Apr 2019.
Smit AFA, Hubley R, Green P: RepeatMasker Open-4.0. 2013-2015 <http://www.repeatmasker.org>. Accessed 22 Apr 2019.
Stanke M, Tzvetkova A, Morgenstern B. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 2006;7(Suppl 1):S11 11–18.
Li H: Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, preprint arXiv:1303.3997.
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31(12):2032–4.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Mayer KF, Waugh R, Brown JW, Schulman A, Langridge P. A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012;491.
Du HL, Yu Y, Ma YF, Gao Q, Cao YH, Chen Z, Ma B, Qi M, Li Y, Zhao XF, et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat Commun. 2017;8.
Jiao YP, Peluso P, Shi JH, Liang T, Stitzer MC, Wang B, Campbell MS, Stein JC, Wei XH, Chin CS, et al. Improved maize reference genome with single-molecule technologies. Nature. 2017;546(7659):524.
Vogel JP, Garvin DF, Mockler TC, Schmutz J, Rokhsar D, Bevan MW, Barry K, Lucas S, Harmon-Smith M, Lail K, et al. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463(7282):763–8.
Maughan PJ, Lee R, Walstead RN, Vickerstaff RJ, Fogarty MC, Brouwer CR, Reid RR, Jay JJ, Bekele WA, Jackson EW et al: Raw sequences used for A. atlantica genome assembly are deposited in the Sequence Read Archive database under the BioProject PRJNA546592. National Center for Biotechnology Information; 2019: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA546592. Accessed 5 Jun 2019.
Maughan PJ, Lee R, Walstead RN, Vickerstaff RJ, Fogarty MC, Brouwer CR, Reid RR, Jay JJ, Bekele WA, Jackson EW et al: Raw sequences used for A. eriantha genome assembly are deposited in the Sequence Read Archive database under the BioProject PRJNA546595. National Center for Biotechnology Information; 2019: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA546595. Accessed 5 Jun 2019.
Maughan PJ, Lee R, Walstead RN, Vickerstaff RJ, Fogarty MC, Brouwer CR, Reid RR, Jay JJ, Bekele WA, Jackson EW et al: The raw reads for the resequencing panel of the Avena diploid species are found in BioProject PRJNA556219. National Center for Biotechnology Information; 2019: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA556219. Accessed 23 Jul 2019.
Maughan PJ, Lee R, Walstead RN, Vickerstaff RJ, Fogarty MC, Brouwer CR, Reid RR, Jay JJ, Bekele WA, Jackson EW et al: Avena atlantica genome and annotation. Comparative Genomics; 2019: https://genomevolution.org/coge/GenomeInfo.pl?gid=53337. Accessed 21 Dec 2018.
Maughan PJ, Lee R, Walstead RN, Vickerstaff RJ, Fogarty MC, Brouwer CR, Reid RR, Jay JJ, Bekele WA, Jackson EW et al: Avena eriantha genome and annotation. Comparative Genomics; 2019: https://genomevolution.org/coge/GenomeInfo.pl?gid=53381. Accessed 2 Jan 2019.
Acknowledgements
We gratefully acknowledge David A. Kudrna (Arizona Genomics Institute, Tucson, AZ, USA) and Edward Wilcox (DNA sequencing Center, Brigham Young University, Provo, UT, USDA) for their assistance and expertise with PacBio sequencing.
Funding
The funding for this research was provided through a grant (#1444575) from the Plant Genome Research Program at the National Science Foundation.
Author information
Authors and Affiliations
Contributions
PJM, ENJ, and JS conceived and designed the study. RL and MC performed the experiments and managed the plant materials. PJM, RW, RJV, CB, RRR, JJ, and WAB performed the bioinformatic analyses, including de novo genomic and transcriptome assemblies, annotations, and SNP discovery. EJ, NAT, and TL contributed to the comparative genomics and candidate gene discovery for heading date and crown rust. PJM, ENJ, and JS wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Table S1. Summary of the repeat element content in the amaranth genome assembly as identified by RepeatMasker relative to the RepBase-derived RepeatMasker libraries.
Additional file 2.
Telomeric satellite and centromeric repeat sequences.
Additional file 3:
Table S2. List of Avena accessions included in the resequencing panel. Cc 7277 (A. atlantica) and CN 19328 (A. eriantha) were the reference genomes. The species and genome formula of each accession is presented. The raw read files can be found in BioProject PRJNA556219.
Additional file 4:
Table S3. SNPs per chromosome use for maximum likelihood phylogeny produced using SNPhylo [73].
Additional file 5:
Figure S1. Rate of synonymous substitutions per synonymous sites (Ks) within duplicated gene pairs from coding sequences predicted from A. atlantica comparisons with A. eriantha, H. vulgare, B. distachyon, O. sativa, and Z. mays.
Additional file 6:
Figure S2. Orthologous genes were identified between A. atlantica and A. eriantha genomes to detect orthologous chromosome relationships. Genome synteny was (A) visualized by dotplot analysis, with boxes drawn around syntenic regions, (B) quantified, where the chromosome pairs with the highest amount of syntenic block connections, expressed as a percentage of the total syntenic bases, are colored red and transition to white as the number of connections decreases and (C) correlation between syntenic block sizes between A. atlantica and A. eriantha.
Additional file 7:
Figure S3. Orthologous genes were identified between A. atlantica and H. vulgare genomes to detect orthologous chromosome relationships. Genome synteny was (A) visualized by dotplot analysis, with boxes drawn around syntenic regions, (B) quantified, where the chromosome pairs with the highest amount of syntenic block connections, expressed as a percentage of the total syntenic bases, are colored red and transition to white as the number of connections decreases and (C) correlation between syntenic block sizes between A. atlantica and H. vulgare (Hv_IBSC_PGSB_v2; Ensembl Release 36).
Additional file 8:
Figure S4. Orthologous genes were identified between A. eriantha and H. vulgare genomes to detect orthologous chromosome relationships. Genome synteny was (A) visualized by dotplot analysis, with boxes drawn around syntenic regions, (B) quantified, where the chromosome pairs with the highest amount of syntenic block connections, expressed as a percentage of the total syntenic bases, are colored red and transition to white as the number of connections decreases and (C) correlation between syntenic block sizes between A. eriantha and H. vulgare (Hv_IBSC_PGSB_v2; Ensembl Release 36).
Additional file 9:
Table S4. Summary of resistance gene analog identification results using the RGAugury pipeline [92] for A. atlantica and A. eriantha.
Additional file 10:
Figure S5. Distribution of the resistance gene analogs (RGAs) encoding genes on the (A) A. atlantica and (B) A. eriantha genome. The RGAugury pipeline classifies RGA candidates into four major families based on the presence of RGA domains and motifs, specifically, nucleotide binding sites (NBS, blue), transmembrane coiled-coil (TMCC, black), and membrane associated receptor-like proteins kinases (RLK, yellow) and receptor-like proteins (RLP, green). The predicted location of the Pc91 crown rust QTLs (Klos et al. [30]) in the A. atlantica genome is shown on chromosome AA2.
Additional file 11:
Table S5. Candidate resistance gene analogs associated with crown rust resistance on Mrg18 linkage group [28]. Mrg18 was previously shown to be involved in an intergenomic translocation involving 7C and 17A, corresponding to A. eriantha chromosome AE7 and A. atlantica chromosome AA2. Klos et al. [30] identified two QTLs associated P. coronata (crown rust) resistance on Mrg18, one of which determined to be Pc91. Candidate resistance gene analogs were identified using BLAST searches against the A. atlantica and A. eriantha genome assembly using makers sequences associated with the QTLs.
Additional file 12:
Figure S6. Corresponding location of restriction fragment length polymorphism markers mapped on a segregating A. strigosa X A. wiestii population developed by Kremer et al. [102] on the A. atlantica chromosomes (shown on a gene density plot). Markers from each of their linkage groups (Asw A-I) are color-coded with approximate positions on A. atlantica scaffolds indicated with arrows.
Additional file 13:
Table S6. PacBio and Illumina sequencing read statistics. The raw read files can be found in BioProjects PRJNA546592 and PRJNA546595.
Additional file 14:
Table S7. Raw read statistics for RNASeq data for A. atlantica and A. eriantha. All reads were illumina pair-end reads from standard 500 bp insert libraries. The raw read files can be found in BioProjects PRJNA556794 and PRJNA546595.
Additional file 15:
Figure S7. Unabbreviated A-genome diploids rooted to the A. eriantha reference (ER_CN 19238). Accession names are abbreviated as described in Additional file 3: Table S2.
Additional file 16:
Figure S8. Unabbreviated C-genome diploids rooted to the A. atlantica (AT_Cc 7277) reference. Accession names are abbreviated as described in Additional file 3: Table S2.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Maughan, P.J., Lee, R., Walstead, R. et al. Genomic insights from the first chromosome-scale assemblies of oat (Avena spp.) diploid species. BMC Biol 17, 92 (2019). https://doi.org/10.1186/s12915-019-0712-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-019-0712-y