
Abstract
Background
Hirudin is an anti-coagulation protein produced by the salivary glands of the medicinal leech Hirudomedicinalis. It is a powerful and specific thrombin inhibitor. The novel recombinant hirudin, RGD-hirudin, which contains an RGD motif, competitively inhibits the binding of fibrinogen to GPIIb/IIIa on platelets, thus inhibiting platelet aggregation while maintaining its anticoagulant activity.
Results
Recombinant RGD-hirudin and six mutant variants (Y3A, S50A, Q53A, D55A, E57A and I59A), designed based on molecular simulations, were expressed in Pichia pastoris. The proteins were refolded and purified to homogeneity as monomers by gel filtration and anion exchange chromatography. The anti-thrombin activity of the six mutants and RGD-hirudin was tested. Further, we evaluated the binding of the mutant variants and RGD-hirudin to thrombin using BIAcore surface plasmon resonance analysis (SPR). Kinetics and affinity constants showed that the KD values of all six mutant proteins were higher than that of RGD-hirudin.
Conclusions
These findings contribute to a novel understanding of the interaction between RGD-hirudin and thrombin.
Similar content being viewed by others
Background
A highly specific thrombin inhibitor derived from the leech (Hirudomedicinalis hirudin) has come into use over the past decade in in vitro blood research. It blocks the thrombin-mediated conversion of fibrinogen to fibrin during clot formation, but unlike heparin, it is a direct thrombin inhibitor (DTI) [1] that is not inactivated by platelet factor 4 (PF4) [2],[3].
A previous study has shown that the inhibitor is a small peptide (65 amino acids, 7 kDa) that binds to active thrombin and irreversibly inactivates it [4]. It has also been thoroughly characterized in several laboratories by biochemical and biophysical means, including determination of nuclear magnetic resonance (NMR) structures in solution of both natural and recombinant variants [5]-[7]. These investigations revealed that hirudin comprises an N-terminal globular domain (residues Ile1-Ile49) stabilized by three disulfide bonds with [1]-[5],[4]-[6] connectivity, which spontaneously folds in solution [8]. This compact domain is extended on the C-terminus by a short acidic tail that lacks cysteine residues and is essentially disordered in solution [9]. Structural studies conducted on hirudin in the free [5]-[7],[10] and thrombin-bound state [11],[12] indicate that both the N-terminus (Tyr3, Asp5) [11],[13] and the C-terminus play an important role in the interaction with thrombin. Notably, the long, extended conformation of the C-terminus interacts with a multitude of residues on the surface of thrombin.
The novel recombinant RGD-hirudin, which contains an Arg-Gly-Asp (RGD) adhesion site recognition sequence, is a bi-functional molecule based on the structure of wild-type hirudin variant 2 [14]. In the recombinant version, several amino acid residues in the C-terminus have been replaced by negatively charged residues (Asp62 and Asp65). Asp53 was mutated to Gln53, Glu58 was mutated to Pro58, and Glu66 was added. These changes improve the hydrophobicity of the protein and allow the recombinant RGD-hirudin to interact more effectively with the fibrinogen recognition exosite of thrombin, resulting in a specific activity of 12,000 ATU/mg [15]. Given these changes, we hypothesized that the interaction between RGD-hirudin and thrombin would be similar to that between wild-type hirudin and thrombin. To test this, we expressed and purified RGD-hirdudin and six mutant variants in Pichia pastoris. The design of the six mutations was based on molecular simulations. The Titration Testing Method and BIAcore® surface plasmon resonance analysis (SPR) were used to test their thrombin inhibition activity. Anti-platelet aggregation activity was tested by classic turbidity assays.
Methods
Materials
Accelrys Discovery Studio (DS, version 3.1) was used for homology modeling (MODELER) [16] and docking simulation (ZDOCK) [17],[18]. Pichia pastoris cells carrying the RGD-hirudin gene (Mut+) and pPIC9k-RGD-hirudin plasmid were stored in our lab. Briefly, the RGD-hirudin gene was synthesized in the Key Laboratory of Molecular Medicine at Fudan University. cDNA encoding RGD-hirudin was cloned into the plasmid pPIC9K, and this expression vector was transformed into Pichia pastoris GS115. Vector integration into the Pichia pastoris chromosome was confirmed by PCR [14],[19]. DNA primers were synthesized by Sangon Biotech (Shanghai) Co., Ltd. The Site-Directed Mutagenesis Kit was purchased from SBS Genetech Co., Ltd. Yeast nitrogen base was obtained from Sigma Aldrich Co., Ltd. Blood plasma was obtained from the Shanghai Blood Center. Sephacryl S-100 HR, Sephadex-G50, and Q-Sepharose-FF were purchased from GE Healthcare Co., Ltd. The Biacore T100 instrument and research grade CM5 chips were purchased from Biacore (GE Healthcare) Co., Ltd. Other reagents were of analytical purity.
Homology modeling
The amino acid sequence of RGD-hirudin was obtained in our lab. The NCBI protein BLAST program was used to search the Protein Data Bank (PDB) and was used to select a template structure for RGD-hirudin homology modeling. From the selected templates, a three-dimensional model of RGD-hirudin was obtained by homology modeling using the software package Discovery Studio 3.1. Constructed models were refined by performing an optimized geometry calculation of the mechanics using augmented CHARMM in Discovery Studio 3.1. The quality of refined models was assessed on the basis of both geometry and energy. The stereo-chemical properties of the models were investigated with a Ramachandran plot using PROCHECK [20].
Protein-protein docking
To assess the interaction between RGD-hirudin and thrombin, docking the modeled structure of RGD-hirudin with the crystal structure of thrombin (PDB ID: 4HTC) chain H&L was performed. ZDOCK tested the different docking sites by moving the ligand around the receptor. To obtain more accurate predictions, we specified the angular step size for the rotational sampling of the ligand orientations as 6, and the shape complementarity, desolvation and electrostatic energy terms were used in the initial-stage ranking of the docked protein poses. The docking parameters are set as follow: Input Receptor Protein: 4HTC: Thrombin, Input Ligand Protein: RGD-Hirudin, Angular Step Size: 6, ZRank: True, Clustering: Top poses: 2000, RMSD Cutoff: 6, Interface Cutoff: 9, Maximum Number of Clusters: 60.
Mutation Energy Calculation (binding affinity)
The Mutation Energy Calculation was used to evaluate the effect of single-point mutations on the binding affinity in the RGD-hirudin-thrombin docking complex. This protocol performs amino acid scanning mutagenesis on a set of residues (selected according to the docking results) by mutating each to alanine. The energy effect on the binding affinity of each mutation (Mutation Energy) was calculated as the difference in the binding free energy between the mutant and wild-type proteins. The binding free energy is defined as the difference in the free energy between the complexed and unbound state. All interaction energy terms were calculated by CHARM using a Generalized Born implicit solvent model; these terms contained empirically scaled contributions of van der Waals and electrostatic interactions and a non-polar solvation energy term. The mutation energy function contained also a side chain entropy term. The temperature was set at 37°C. This process was also implemented with the software package Discovery Studio 3.1.
Cloning, expression, and purification of RGD-hirudin and the mutant variants
For preparation of six RGD-hirudin mutants, a Site-Directed Mutagenesis Kit was using two complementary primers which were described as follows (Table 1) and the pPIC9k-RGD-hirudin plasmid as a template. The procedure consisted of 18 polymerase chain reaction cycles as specified by the manufacturer’s manual using Pfu polymerase. The mutated construct was then digested with the restriction enzyme DpnI, which is specific to methylated and hemi-methylated DNA, to digest the template and select mutation-containing newly-synthesized DNA. DNA sequencing confirmed that all six mutant plasmids were correct.
Wild type and mutant variants of RGD-hirudin were expressed in Pichia pastoris, then purified by gel filtration and anion exchange chromatography as previously described [14],[21]. Briefly, a single colony was used to inoculate 5 mL of YPD in a 50 mL flask, which was incubated for 14 h at 30°C at 220 rpm. This culture was used to inoculate 200 mL of sterile BMGY. Cultures were incubated at 30°C and 220 rpm for 30 h to an OD600 of 5.8. Cells were harvested by centrifugation at 3000 rpm for 30 min, washed with 100 mM potassium phosphate (pH 6.0), and carefully resuspended in 200 mL sterile BMM. Samples were incubated at 30°C for another 72 h, with pulses of 0.5% (v/v) methanol added every 12 h. The culture was centrifuged and the supernatant was loaded onto a Sephacryl-S100 column (7.5 cm × 80 cm), pre-equilibrated with 20mMol/L phosphate buffer (PB, pH 7.4). A sample of 400 mL was eluted from the gel filtration column and loaded onto a Q-Sepharose FF column (2.6 cm × 20 cm), also pre-equilibrated with 20mMol/L PB (pH 7.4). The column was washed with 20mMol/L PB (pH 7.4), followed by a single linear gradient of 0–1.0Mol/L NaCl-PB buffer. RGD-hirudin was eluted at 0.25Mol/L NaCl-PB. The sample that showed anti-thrombin activity was collected and desalted with a Sephadex-G50 column (1.6 cm × 20 cm). Loading sample volumes were standardized to 5 mL. Protein concentration was measured with the Bradford assay. The desalted samples were lyophilized and stored at −80°C.
Protein identification
Protein samples (wild type and mutant variants of RGD-hirudin) were analyzed by 15% SDS-PAGE. The anti-thrombin activity of RGD-hirudin and the mutant variants was tested by the Titration Testing Method, according to Markwardt [22]. Briefly, 200 mL of fresh plasma was added to a 1.5 mL tube. A 5 mL sample of RGD-hirudin was added to the plasma and mixed by vortexing. Thrombin (5 mL of 100 NIH units) was added and allowed to stand for 1 min at 37°C. If the plasma did not clot, the RGD-hirudin contained 100 units of anti-thrombin activity. Thus, consumption of 1 NIH unit of thrombin is equivalent to 1 unit of anti-thrombin activity. A classic turbidity assay was used to measure the anti-platelet aggregation activity of RGD-hirudin and the mutant variants [15]. Aggregation of rabit platelets in response to antagonists was analyzed using lumi-aggregometer (Model 400VS, Chrono-Log, Haverston, PA, USA).
Surface plasmon resonance interaction analysis (BIAcore)
Thrombin was diluted in 5mMol/L sodium acetate (pH 5.0), and 2000 response units were immobilized via amine coupling to CM5 sensor chip flow chambers (GE Healthcare). Briefly, proteins were mixed with equal volumes of freshly prepared 100mMol/L N-hydroxysuccinimide and 400mMol/L N-ethyl-N’-(dimethylaminopropyl) carbodiimide, and capping of unreacted carboxymethyl sites was achieved by injection of 1.0Mol/L ethanolamine, pH 7.0. A flow chamber was subjected to the immobilization protocol. RGD-hirudin and the six mutant variants were sequentially diluted in running buffer (10mMol/L HEPES, 150mMol/L NaCl, 0.005% Surfactant P20 (BIAcore), pH 7.5) and injected over the surfaces at different concentrations (RDG-hirudin: 0-50nMol/L; Y3A: 0-10mMol/L; S50A: 0-50nMol/L; Q53A: 0-5μMol/L; D55A: 0-10μMol/L; E57A: 0-10μMol/L; I59A: 0-50μMol/L) at 30 μL/min.
Binding was monitored with a BIAcore T100 instrument. Between experiments, the surfaces were strictly regenerated with multiple pulses of 2Mol/L NaCl and 1.5Mol/L glycine-HCl, pH 2.5, followed by an extensive wash procedure with running buffer.
After x- and y-axis normalization of the data obtained, the blank bulk refraction curves from the control flow chamber of each injected concentration were subtracted. Binding curves were displayed, and the association (Ka) and dissociation (Kd) rate constants were determined using the BIAevaluation 4.1 software and its equation for 1:1 Langmuir binding. From these values, affinities (KD) were calculated.
Results
Molecular modeling of RGD-hirudin
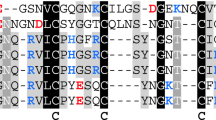
RGD-hirudin was molecularly modeled, based on the crystal structure of hirudin variant 2 from chain I of the hirudin-thrombin complex (PDB ID: 4HTC) [23] and the NMR solution structure of chain A of recombinant RGD-hirudin (PDB ID: 2JOO) [24]. Chain I shares 80% and chain A shares 100% sequence identity with RGD-hirudin. Residues 52–66 of the structure, which may play important roles in the interaction with thrombin, were absent. The sequence alignment is shown in Figure 1A. The three-dimensional structural model for RGD-hirudin was generated based on the 4HTC and 2JOO templates using the Discovery Studio 3.1 software.

Molecule simulation of RGD-hirudin. (A) Amino acid sequence alignment of RGD-hirudin, chain I of hirudin variant 2 from the hirudin-thrombin complex (PDB ID: 4HTC) and the NMR solution structure of chain A of recombinant RGD-hirudin (PDB ID: 2JOO). (B) Ramachandran plot of the selected RGD-hirudin model. The distribution of the RGD-hirudin residues (black dots) are shown in color: most favorable (red), additional allowed (yellow), generously allowed (light yellow), and disallowed (white) regions. (C) Homology modeling of the RGD-hirudin structure.
Analysis of the Ramachandran plot of the refined model showed that 96.9% of the residues lie in the most favorable regions (Figure 1B), which indicates that the model is suitable for structural studies (Figure 1C).
Thrombin docked with RGD-hirudin
The docking simulation of RGD-hirudin and thrombin resulted in 54000 poses for analysis. The poses were clustered with the cluster root-mean-square deviation (RMSD) cutoff and the interface cutoff both set to 10 angstroms. For a pose to be included in a cluster, the RMSD cutoff was the maximal RMSD of the ligand interface from the cluster center, and the interface cutoff was the size of the interface region between receptor and ligand. Poses were then re-ranked with Van Der Waals, desolvation and electrostatics energy terms. The best docking poses were selected, based on a higher dock score combined with certain types of interactions between hirudin variant 2 and thrombin. We hypothesized that the interaction between RGD-hirudin and thrombin is similar to that of hirudin variant 2 and thrombin, and therefore favored this type of interaction. Finally, pose 24 was chosen as the best docking complex (Figure 2A). A close examination of the region in the functional site of RGD-hirudin when in complex with thrombin revealed that Tyr3 in the N-terminus of RGD-hirudin formed one hydrogen bond with Gly210 of thrombin. Further, in the C-terminus of RGD-hirudin, Ser50 formed two hydrogen bonds with Glu192 of thrombin, Gln53 formed one hydrogen bond with Leu40 of thrombin, Asp55 formed two hydrogen bonds with Arg73 of thrombin, and both Glu57 and Ile59 were hydrogen-bonded to Gln38 of thrombin (Figure 2B).

Thrombin docked with RGD-hirudin. Thrombin is represented by its solvent-accessible surface, while hirudin is shown as a solid ribbon. (A) The best docking complex of RGD-hirudin and thrombin (pose 24). (B) Close-up of the region of the RGD-hirudin functional site in complex with thrombin. Residues of RGD-hirudin are in yellow. Residues of thrombin are in blue. Hydrogen bond interactions are indicated with dashed green lines.
Calculating the effect of single-point mutations on binding affinity
We performed amino acid scanning mutagenesis of RGD-hirudin-thrombin complexes, mutating to alanine each of six residues shown by the docking results to be important. We then calculated the differences in the free energy of binding between the wild-type and mutated structures. The results showed that the binding affinity of the six single-point mutations with thrombin were weaker (weighted mutation energy greater than zero) than that of wild-type RGD-hirudin with thrombin. The effect of each mutation was destabilizing (Table 2), which further confirmed the docking results and demonstrates that the six residues (Tyr3, Ser50, Gln53, Asp55, Glu57, Ile59) of RGD-hirudin might participate in the interaction with thrombin.
Purification and characterization of RGD-hirudin and mutant variants
In this study, we focused on the residues of RGD-hirudin that are hydrogen-bonded to thrombin. These six residues (Tyr3, Ser50, Gln53, Asp55, Glu57, Ile59) were mutated to Ala, and the proteins were purified by gel filtration and anion exchange chromatography, as described in the Materials and Methods. The average yields of pure protein from 200 mL of fermentation culture were: RGD-hirudin, 46 mg; Y3A, 50 mg; S50A, 42 mg; Q53A, 47 mg; D55A, 40 mg; E57A, 38 mg; and I59A, 55 mg (Figure 3A).

Expression, purification and characteristic of RGD-hirudin and six mutant versions. (A) Expression and purification of RGD-hirudin and six mutant versions were analyzed by 15% SDS-PAGE. Lane M: molecular mass standards (116 to 14.4 kDa) 20 μL (1 mg/mL) of each sample was loaded on the gel. (B) Anti-thrombin activity of purified RGD-hirudin and six mutant variants. (C) RGD-hirudin and the six mutants have similar effects on rabbit platelet aggregation induced by ADP. The experiments were performed five times with similar results. Error bars represent the standard deviation.
The anti-thrombin specific activity of the purified RGD-hirudin was almost 18000 ATU/mg, while the mutated versions had activities reduced to different degrees: Y3A, 4000 Anti-thrombin Units (ATU)/mg; S50A, 16000 ATU/mg; Q53A, 14000 ATU/mg; D55A, 12000 ATU/mg; E57A, 11000 ATU/mg; and I59A, 9000 ATU/mg (Figure 3B). The dramatic decline in anti-thrombin specific activity of the Y3A mutation suggests that Tyr3 in RGD-hirudin might be a very important residue for its interaction with thrombin.
Anti-platelet aggregation activities of the proteins were determined by a classic turbidity assay. The results showed there was no significant difference between RGD-hirudin and the mutant variants (Figure 3C).
SPR measurement of the interactions of RGD-hirudin and six mutant variants with thrombin
To further analyze the binding properties of the six mutant variants of RGD-hirudin with thrombin and to establish the affinity constant of the binding, surface plasmon resonance analysis was performed. The binding activity of RGD-hirudin was compared with those of the six mutants (Y3A, S50A, Q53A, D55A, E57A and I59A) in a competitive non-radioactive binding assay using thrombin as a receptor. The KD (affinity) was calculated from the determined Ka and Kd values (Table 3). The KD for the interaction between RGD-hirudin and thrombin was 15.5nM (Figure 4A), whereas the KD of thrombin binding for the mutant Y3A was 3.6 mM; S50A, 35nM; Q53A, 0.57 μM; D55A, 1 μM; E57A, 2.4 μM; and I59A, 19 μM (Figure 4B-G). BIAcore analysis thus confirmed that the binding affinity of thrombin and wild-type RGD-hirudin is slightly stronger than that of the S50A mutant, and much stronger than those of the other five variants, especially Y3A. Meanwhile, the affinity curves of RGD-hirudin and the six mutants (at the same molarity of 100nM) and thrombin demonstrated that the binding affinities for thrombin of all six mutant versions were weaker than that of RGD-hirudin (Figure 4H). In conclusion, the six residues (Tyr3, Ser50, Gln53, Asp55, Glu57 and Ile59) are important for binding to thrombin, as each mutant protein binds with lower affinity than wild-type RGD-hirudin.

SPR measurement of the interactions of RGD-hirudin and mutant variants with thrombin. (A-G) KD values. BIAcore analysis showed that the binding affinity of thrombin and wild-type RGD-hirudin was slightly stronger than those of the six mutants. (H) Affinity curves (with proteins at 100nM) demonstrated that the binding affinities of thrombin and the six mutants were weaker than that of RGD-hirudin.
Discussion
Hirudin, an antithrombotic substance produced by the salivary glands of the medicinal leech (Hirudomedicinalis) [25],[26], is the most potent and specific thrombin inhibitor currently known. It acts by binding directly via multiple sites to thrombin: the N-terminal globular domain binds near to the active site of thrombin, while the extended C-terminal segment, which is abundant in acidic residues and includes a sulfated tyrosine, has both ionic and hydrophobic interactions with the thrombin fibrinogen recognition exosite (FRE) [23].
In our laboratory, a new type of recombinant RGD-hirudin has been successfully cloned, expressed in the methylotrophic yeast Pichia pastoris and subsequently purified. It may be a more effective agent than wild-type hirudin for anti-coagulation and anti-thrombosis in post-anastomosis surgery. We propose that RGD-hirudin will be able to replace heparin or wild-type hirudin and reduce bleeding rates, due to its bi-functional action at a much lower dosage than wild-type hirudin [15]. However, RGD-hirudin needed to be modified to reduce the molecular weight to be suitable for advanced oral administration, rather than intravenous injection.
Although our lab has previously conducted a study on the structure of RGD-hirudin in solution (1–49) [24], the structure of the extended C-terminal domain (from Asn52 to Glu66), which is essential for the hirudin-thrombin interaction, had not yet been determined. The overall structure of RGD-hirudin (1–66) developed above used the crystal structure of chain I of hirudin variant 2 from the hirudin-thrombin complex (PDB ID: 4HTC) and the NMR solution structure of chain A of recombinant RGD-hirudin (PDB ID: 2JOO) as templates for docking with thrombin. Based on the docking model and the structural characteristics of the RGD-hirudin-thrombin complex, we find that the mechanism of interaction is consistent with that of native hirudin and thrombin. In this interaction, the globular N-terminal domains interact with the active site of thrombin, while the anionic C-terminal tails bind to exosite 1 on thrombin (the substrate-binding site) [27]. Six residues (Tyr3, Ser50, Gln53, Asp55, Glu57 and Ile59) of RGD-hirudin are hydrogen-bonded to thrombin, according to the results of simulations. To validate these residues as functional sites, mutant variants were constructed. Anti-thrombin activity assays and surface plasmon resonance analysis were also performed. Previous studies have indicated that the first three residues in the N-terminus of hirudin represent about 30% of the binding energy [28] and the acidic C-terminal segment accounts for about 32% of the binding energy at zero ionic strength [29]. Mutagenesis studies have shown that all the negatively charged residues of the C-terminal tail are relevant for complex formation [30]-[32]. Our results imply that RDG-hirudin’s N-terminus (Tyr3) and C-terminus (Ser50, Gln53, Asp55, Glu57 and Ile59) are both crucial for inhibiting the activity of thrombin, for binding near its active site and forming hydrogen bonds with exosite I.
In this study, we focused on hydrogen bonding interactions between RGD-hirudin and thrombin. However, hydrophobic interactions, Van der Waals interactions, and salt bridges might be also exist. For example, the phenyl ring of Phe56 engages in strong Van der Waals, edge-to-face interactions with thrombin Phe34, according to our docking complex (data not shown). Thus, other mechanisms of interacting and binding should be investigated in the future.
Conclusions
In summary, The purpose of this study is that we found the binding site and active centre of RGD-Hirduin. In future, these results might be useful to provide a structural basis for the redesign of lower molecular weight antithrombin peptides which would be developed to the new oral drugs to fight thrombotic diseases.
Abbreviations
- RGD:
-
Arginine-Glycine-Aspartic acid
- SPR:
-
Surface plasmon resonance analysis
- DTI:
-
Direct thrombin inhibitor
- PF4:
-
Platelet factor 4
- NMR:
-
Nuclear magnetic resonance
- PB:
-
Phosphate buffer
- Ka:
-
Association rate constants
- Kd:
-
Dissociationrate constants
- KD:
-
Affinities constants
- RMSD:
-
Root-mean-square deviation
- ATU:
-
Anti-thrombin units
- FRE:
-
Fibrinogen recognition exosite
References
Sun Z, Zhao Z, Zhao S, Sheng Y, Zhao Z, Gao C, et al.: Recombinant hirudin treatment modulates aquaporin-4 and aquaporin-9 expression after intracerebral hemorrhage in vivo. Mol Biol Rep 2009, 36: 1119–1127. 10.1007/s11033-008-9287-3
Greinacher A, Pötzsch B, Amiral J, Dummel V, Eichner A, Mueller-Eckhardt C: Heparin-associated thrombocytopenia: isolation of the antibody and characterization of a multimolecular PF4-heparin complex as the major antigen. Thromb Haemost 1994, 71: 247–251.
Amiral J, Bridey F, Dreyfus M, Vissoc AM, Fressinaud E, Wolf M, Meyer D: Platelet factor 4 complexed to heparin is the target for antibodies generated in heparin-induced thrombocytopenia. Thromb Haemost 1992, 68: 95–96.
Chang JY: The functional domain of hirudin, a thrombin-specific inhibitor. Febs Lett 1983, 164: 307–313. 10.1016/0014-5793(83)80307-X
Clore GM, Sukumaran DK, Nilges M, Zarbock J, Gronenborn AM: The conformations of hirudin in solution: a study using nuclear magnetic resonance, distance geometry and restrained molecular dynamics. Embo J 1987, 6: 529–537.
Folkers PJ, Clore GM, Driscoll PC, Dodt J, Köhler S, Gronenborn AM: Solution structure of recombinant hirudin and the Lys-47-Glu mutant: a nuclear magnetic resonance and hybrid distance geometry-dynamical simulated annealing study. Biochemistry 1989, 28: 2601–2617. 10.1021/bi00432a038
Haruyama H, Wuthrich K: Conformation of recombinant desulfatohirudin in aqueous solution determined by nuclear magnetic resonance. Biochemistry 1989, 28: 4301–4312. 10.1021/bi00436a027
Chatrenet B, Chang JY: The disulfide folding pathway of hirudin elucidated by stop/go folding experiments. J Biol Chem 1993, 268: 20988–20996.
Corral-Rodriguez MA, Macedo-Ribeiro S, Pereira PJ, Fuentes-Prior P: Leech-derived thrombin inhibitors: from structures to mechanisms to clinical applications. J Med Chem 2010, 53: 3847–3861. 10.1021/jm901743x
Szyperski T, Guntert P, Stone SR, Tulinsky A, Bode W, Huber R, et al.: Impact of protein-protein contacts on the conformation of thrombin-bound hirudin studied by comparison with the nuclear magnetic resonance solution structure of hirudin(1–51). J Mol Biol 1992, 228: 1206–1211. 10.1016/0022-2836(92)90326-F
Rydel TJ, Tulinsky A, Bode W, Huber R: Refined structure of the hirudin-thrombin complex. J Mol Biol 1991, 221: 583–601. 10.1016/0022-2836(91)80074-5
Grutter MG, Priestle JP, Rahuel J, Grossenbacher H, Bode W, Hofsteenge J, et al.: Crystal structure of the thrombin-hirudin complex: a novel mode of serine protease inhibition. Embo J 1990, 9: 2361–2365.
Skrzypczak-Jankun E, Carperos VE, Ravichandran KG, Tulinsky A, Westbrook M, Maraganore JM: Structure of the hirugen and hirulog 1 complexes of alpha-thrombin. J, Mol, Biol 1991, 221: 1379–1393. 10.1016/0022-2836(91)90939-4
Huang YN, Zhang YL, Wu Y, Wang J, Yu M, Mo W, et al.: Expression, purification, and mass spectrometric analysis of (15)N, (13)C-labeled RGD-hirudin, expressed in pichia pastoris, for NMR studies. PLoS One 2012, 7: e42207. 10.1371/journal.pone.0042207
Mo W, Zhang YL, Chen HS, Wang LS, Song HY: A novel hirudin derivative characterized with anti-platelet aggregations and thrombin inhibition. J Thromb Thrombolysis 2009, 28: 230–237. 10.1007/s11239-008-0251-9
Sali A, Blundell TL: Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993, 234: 779–815. 10.1006/jmbi.1993.1626
Chen R, Li L, Weng Z: ZDOCK: an initial-stage protein-docking algorithm. Proteins 2003, 52: 80–87. 10.1002/prot.10389
Chen R, Weng Z: A novel shape complementarity scoring function for protein-protein docking. Proteins 2003, 51: 397–408. 10.1002/prot.10334
Mo W, Zhang YL, Wang LS, Yang XY, Song HY: Fermentation, purification and identification of recombinant RGD-hirudin. Chin J Biotechnol 2004, 20: 126–129.
Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM: AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 1996, 8: 477–486. 10.1007/BF00228148
Wang J, Zhang YL, Song HY, Yu M, Dai LS, Mo W, et al.: Overproduction of 15 N-labeled r-RGD-hirudin in pichia pastoris for NMR studies. Protein Pept Lett 2010, 17: 1228–1233. 10.2174/092986610792231465
Markwardt F, Walsmann P: Purification and analysis of the thrombin inhibitor hirudin. Hoppe Seylers Z Physiol Chem 1967, 348: 1381–1386. 10.1515/bchm2.1967.348.1.1381
Rydel TJ, Ravichandran KG, Tulinsky A, Bode W, Huber R, Roitsch C, et al.: The structure of a complex of recombinant hirudin and human alpha-thrombin. Science 1990, 249: 277–280. 10.1126/science.2374926
Song X, Mo W, Liu XG, Yan XM, Song HY, Dai LS, et al.: The NMR solution structure of recombinant RGD-hirudin. Biochem Biophys Res Commun 2007, 360: 103–108. 10.1016/j.bbrc.2007.06.014
Markwardt F. Hirudin: the promising antithrombotic. Cardiovasc Drug Rev. 1992;211–32.
Markwardt F: The development of hirudin as an antithrombotic drug. Thromb Res 1994, 74: 1–23. 10.1016/0049-3848(94)90032-9
Wallis RB: Hirudins: from leeches to man. Semin Thromb Hemost 1996, 22: 185–196. 10.1055/s-2007-999007
Betz A, Hofsteenge J, Stone SR: Interaction of the N-terminal region of hirudin with the active-site cleft of thrombin. Biochemistry 1992, 31: 4557–4562. 10.1021/bi00134a004
Stone SR, Dennis S, Hofsteenge J: Quantitative evaluation of the contribution of ionic interactions to the formation of the thrombin-hirudin complex. Biochemistry 1989, 28: 6857–6863. 10.1021/bi00443a012
Braun PJ, Dennis S, Hofsteenge J, Stone SR: Use of site-directed mutagenesis to investigate the basis for the specificity of hirudin. Biochemistry 1988, 27: 6517–6522. 10.1021/bi00417a048
Myles T, Le Bonniec BF, Betz A, Stone SR: Electrostatic steering and ionic tethering in the formation of thrombin-hirudin complexes: the role of the thrombin anion-binding exosite-I. Biochemistry 2001, 40: 4972–4979. 10.1021/bi0023549
Betz A, Hofsteenge J, Stone SR: Ionic interactions in the formation of the thrombin-hirudin complex. Biochem J 1991, 275(Pt 3):801–803.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (NSFC 21242009, NSFCJ121041, MDJH2012004 and JYH6273102/007/016/003).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HY, ZB, XQ ZX, performed protein expression and purification; ZY constructed the RGD-hirudin expression plasmid and analyzed anti-platelet aggregation activity; HY and MW designed experiments, analyzed data, and wrote the paper; YM is the supervisor of HY. HS is the first inventor of RGD-hirudin. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Huang, Y., Zhang, Y., Zhao, B. et al. Structural basis of RGD-hirudin binding to thrombin: Tyr3 and five C-terminal residues are crucial for inhibiting thrombin activity. BMC Struct Biol 14, 26 (2014). https://doi.org/10.1186/s12900-014-0026-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12900-014-0026-9