
Abstract
Background
Bladder cancer (BC) seriously endangers public health, but effective biomarkers for BC diagnosis, particularly in the early stage, are still lacking. Identification of reliable biomarkers associated with early-stage BC is of great importance to early treatment and an improved outcome.
Methods
Differentially expressed genes (DEGs) were identified using four publicly available early-stage BC gene-expression profiles. Protein–protein interaction (PPI) and survival analysis for hub genes was evaluated. The correlation between methylation of genes and prognosis was evaluated using the MethSurv database. Co-expressed genes were explored using Cancer Cell Line Encyclopedia database and the corresponding expression were assessed in vitro. The competing endogenous RNA network and the immune cell infiltration in BC were generated using data of The Cancer Genome Atlas.
Results
Ten hub genes of the 213 integrated DEGs were identified, including CDH1, IGFBP3, PPARG, SDC1, EPCAM, ACTA2, COL3A1, TPM1, ACTC1, and ACTN1. CDH1 appeared to increase from tumor initiation stage and negatively correlated with methylation. Six methylated sites in CDH1 indicated a good prognosis and one site indicated an aberrant prognosis. High CDH1 expression was negatively correlated with infiltrations by most immune cells, such as plasmacytoid dendritic cells (pDCs), regulatory T cells, macrophages, neutrophils, DCs, and natural killer cells. CDH1 was highly positively correlated with EPCAM and appeared to be directly regulated by miR-383.
Conclusions
The identified oncogenic alterations provide theoretical support for the development of novel biomarkers to advance early-stage BC diagnosis and personalized therapy.
Similar content being viewed by others

Background
Bladder cancer (BC) is one of the most common malignancies worldwide. For decades, BC has had the leading incidence and mortality rates among cancers of the genitourinary system in China [1], posing a serious threat to human health. Among the patients with bladder malignancies, approximately 75% of the diagnoses are related to non–muscle-invasive bladder cancer (NMIBC) at initial presentation [2]. NMIBC is more common in patients under 40 years of age [3], and although patients can be effectively treated by transurethral resection of the bladder tumor and postoperative bladder perfusion chemotherapy [4], NMIBC regularly becomes recurrent, causing fatigue in most patients. In approximately 30% of the patients, NMIBC eventually progresses to muscle-invasive bladder cancer (MIBC) within 5 years [5]. Patients with MIBC have a poor prognosis with a 5-year survival rate of approximately 50% [6].
For patients with BC in the early stage, effective early detection is the key to improving the cure rate and preventing progression to muscle invasion. Currently, cystoscopy and urine cytology are the gold standard for diagnosing BC. However, since relatively frequent cystoscopy involves an invasive examination, it presents a great challenge to the patients’ physical and mental health. Moreover, the sensitivity of urine cytology to low-grade tumors is low [7]. Therefore, more efficient, accurate, and less invasive examination methods have been pursued, and the identification of novel biomarkers for BC has attracted considerable research attention in recent years.
Recently, an increasing number of microarray and high throughput sequencing technologies have been developed to identify biomarkers associated with malignancy, which enable the early diagnosis, prognosis, recurrence monitoring, and exploration of novel drug targets [8]. Integrated bioinformatics technologies have proved effective in overcoming inconsistent results obtained from different platforms and limited cancer sample sizes, facilitating the discovery of a wealth of valuable biological insights [9].
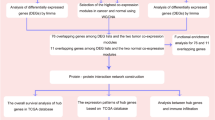
In this study, we identified differentially expressed genes (DEGs) in four raw gene chip expression profile datasets downloaded from the Gene Expression Omnibus (GEO) database, including 29 normal bladder tissue samples and 39 early-stage BC samples (Ta and T1 stage). Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed. A protein–protein interaction (PPI) network was constructed to identify the final hub genes, and the expression of individual genes was assessed by quantitative reverse-transcription polymerase chain reaction (qRT-PCR). The association of specific gene expressions with clinicopathological characteristics and immune infiltration and the correlation between the specific gene methylation and prognosis in BC were further analyzed. Finally, the potential regulators of hub genes were evaluated using a competing endogenous RNA (ceRNA) network. The present study provides promising new insights into potentially reliable biomarkers for tumorigenesis of bladder cancer.
Methods
Microarray data
The GEO database (http://www.ncbi.nlm.nih.gov/geo) [10] is a public genomic data repository that allows users to access high-throughput gene expression data submitted by global research institutions. Four gene expression datasets containing human NMIBC and corresponding adjacent normal tissues [GSE3167 [11], GSE7476 [12], GSE40355 [13], and GSE65635 [14]] were downloaded from GEO. The annotation information on the platform was used as a reference for the corresponding gene symbols. The clinical information of patients with BC and their corresponding mRNA, lncRNA, and miRNA expression data were downloaded from The Cancer Genome Atlas (TCGA) website (https://cancergenome.nih.gov/), as of August 15, 2020. The expression of hub genes in BC and normal bladder samples was evaluated and visualized by the Gene Expression Profiling Interactive Analysis (GEPIA) platform (http://gepia.cancer-pku.cn) [15]. Additionally, RNA-Seq data of candidate genes in different urinary tract cancer cell lines (n = 25) were extracted from the Cancer Cell Line Encyclopedia (CCLE) database (https://portals.broadinstitute.org/ccle/about) [16], as of August 20, 2020.
Integrated analysis of microarray datasets
The limma package [17] in the R/Bioconductor software was used to normalize the matrix data, perform Log2 conversion, and identify DEGs in each microarray dataset. DEGs were integrated using the RobustRankAggreg (RRA) method [18], assuming that each gene ranked randomly in each dataset. If the gene ranked higher across all datasets, the associated P-value was then lower, and the possibility of differential gene expression was greater. A difference in gene expression was considered significant if | Log2FC (fold change) |≥ 1 and adjusted P-value < 0.05.
Function and pathway enrichment analysis
We used the DOSE [19] and clusterProfiler [20] packages of the statistical software R (Version 3.6.2) for mining information related to the biological effects of DEGs and for implementing GO classification and KEGG pathway enrichment [21,22,23]. High quality graphs were displayed using the ggplot2 and pROC packages. Gene set enrichment analysis (GSEA) is a computational method that determines whether an a priori defined set of genes shows significant, concordant differences between two biological states [24]. Gene set enrichment was analyzed using GSEA (version 4.0.3). The functional gene set file ‘c2.cp.kegg.v7.0.symbols.gmt’ summarizes specific and well-defined signaling. The number of substitutions per analysis was set at 1,000, and gene sets with P < 0.05 were recognized as significantly enriched.
PPI network construction and module analysis
An initial PPI network was constructed using the Search Tool for the Retrieval of Interacting Genes (STRING) (version 11.0; http://string-db.org) platform [25]. The minimum value for the highest confidence was set to 0.7, and unconnected proteins were removed from the network. A given network was clustered based on topology using Molecular Complex Detection (MCODE) (version 1.4.2), a plugin of Cytoscape (version 3.4.0), to identify densely connected regions [26]. The final PPI networks were mapped using the Cytoscape visualization software, and the most significant module according to MCODE was identified. The data were filtered based on the following criteria: MCODE score > 5, maximum depth = 100, node score cut-off = 0.2, degree cut-off = 2, and k-score = 2.
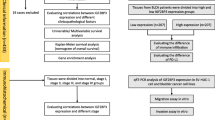
Cell culture
The human BC cell lines 5637 and RT4 and the normal urothelial cell line SVHUC1 were purchased from the Cell Resource Center of the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). All cell lines were cultured in RPMI 1640 medium with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum at 5% CO2 in a 37 ℃ humidified culture environment. Short-tandem repeat profiling was used to authenticate the cell lines less than 6 months before this project was initiated, and the cells were not in culture for more than 2 months.
Analysis of CDH1 methylation and prognosis
CDH1 methylation data was obtained from the cBioPortal (https://www.cbioportal.org/) platform. The correlation between CDH1 methylation and expression level was tested by Spearman correlation analysis and visualized in the cBioPortal platform. Moreover, the prognostic value of the CDH1 methylation level in BC and identification of methylation sites associated with prognosis were evaluated using the MethSurv database (https://biit.cs.ut.ee/methsurv/) [27], which provides a visualization tool to perform survival analysis based on the DNA methylation level of specific genes using TCGA-BLCA data.
Immune infiltration analysis
Immune infiltration levels were evaluated by single-sample GSEA (ssGSEA) using the “GSVA” R package, which can determine the immune cell population in a tumor sample according to gene expression data [28]. The infiltration enrichment of 24 common immune cell types was computed, including B cells, cytotoxic cells, dendritic cells (DCs), activated DCs (aDCs), immature DCs (iDCs), plasmacytoid DCs (pDCs), eosinophils, macrophages, mast cells, neutrophils, natural killer (NK) cells, NK 56- cells, and NK 56 + cells, T cells, CD8 + T cells, T central memory cells (Tcm), T effector memory cells (Tem), T follicular helper cells (Tfh), T gamma delta cells (Tgd), T helper cells (Th), regulatory T cells (Treg), type 1 Th cells (Th1), type 2 Th cells (Th2), and type 17 Th cells (Th17). The correlation between CDH1 expression and immune cell infiltration in BC was evaluated by Spearman rank correlation analysis. The ggplot2 package in the R language was used to show plots of immune cell types. The infiltration level in each immune cell was compared between low and high CDH1 expression groups via Wilcoxon rank sum tests.
RNA isolation and qRT-PCR
Total RNA from each cell line was isolated using TRIzol reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. qRT-PCR was performed using the SYBR Premix Ex Taq II (Perfect Real Time) kit (TaKaRa Bio, Shiga, Japan), with the following settings: 95 °C for 30 s and 39 cycles of 95 ℃ for 5 s and 60 °C for 30 s. DNA dissociation analysis (melting curve) was performed at the end of each run to detect primer dimers, mixed-amplicon populations, and nonspecific products. The relative expression of genes was presented as comparative threshold cycle (2−ΔΔCt) values from at least three independent experiments. The expression of target genes was standardized against Actin Beta (β-actin). The following primer sequences were used: Human CDH1, forward 5′-GAGGCTAACGTCGTAATCACCACA-3′ and reverse 5′-CAAATTGTCCACCATCATCATTCAA-3′; human EPCAM, forward 5′-TGCCAGTGTACTTCAGTTGGT-3′ and reverse 5′-AAAGCCCATCATTGTTCTGGA-3′; and β-actin, forward 5′-AAACGTGCTGCTGACCGAG-3′ and reverse 5′-TAGCACAGCCTGGATAGCAAC-3′.
The ceRNA network construction
To characterize potential regulators of hub genes, a ceRNA network was established. Potential miRNA associated with hub genes was first identified via the TargetScan (http://www.targetscan.org/) and miRDB (http://www.mirdb.org/mmiRDB/) databases. Then, to ensure the quantity of obtained data and matches, differentially expressed lncRNAs (DElncRNAs) (| Log2FC |> 2.0) and DEmiRNA (| Log2FC |> 1.0) with an adjusted P < 0.05 were identified using the edgeR package in R software. Next, miRNAs related to DElncRNAs were predicted using miRcode (http://www.mircode.org/), and miRNAs irrelevant to the hub genes were removed from the network. Finally, these data were integrated, and the ceRNA network was visualized using Cytoscape.
Statistical analysis
The chi-square test was used to analyze the relationship between gene expression and clinical data. The Kaplan–Meier curve and log-rank test were used to plot survival curves. Univariate Cox analysis was used to select relevant variables, and subsequently, multivariate Cox analysis was used for prognostic analysis of gene expression relative to the overall survival (OS) rate of patients with BC. Using the expression level of each mRNA and the regression coefficient obtained from multivariate Cox analysis, a risk score was calculated using the function, Risk score = ExpmRNA1 × βmRNA1 + ExpmRNA2 × βmRNA2 + … + ExpmRNAn × βmRNAn, where Exp represents the expression level of each mRNA and β represents the regression coefficient of each mRNA. An optimal risk model was generated based on the Akaike Information Criterion (AIC) [29]. Patients were divided by median value of risk scores into high-risk and low-risk groups. The predicted power of the prognostic gene signature was determined by the area under the curve (AUC) of the receiver operating characteristic (ROC) curves. P < 0.05 was considered statistically significant.
Results
Identification and integration of DEGs
Thirty-nine NMIBC (Ta and T1 stage) and twenty-nine normal BC samples were enrolled in this study (Table 1). The corresponding clinical and pathological information for each dataset is shown in Additional file 2: Table S1. After normalizing the expression of genes in the four microarray datasets (Additional file 1: Fig. S1), 218, 298, 2925, and 855 up-regulated DEGs and 315, 872, 2399, and 1035 down-regulated DEGs were identified in the GSE3176, GSE7476, GSE40355, and GSE65635 datasets, respectively (Fig. 1A–D). Through integrated analysis by the RRA method, a total of 213 DEGs, 62 up-regulated and 151 down-regulated genes, were finally identified (Additional file 2: Table S2). The top 20 up-regulated and down-regulated genes ranked by fold change are displayed in a heat map (Fig. 1E).

Identification and integration of differentially expressed genes (DEGs). Volcano plots of A GSE3167, B GSE7476, C GSE40355, and D GSE65635, and E heat map of differentially expressed genes. The abscissa represents the GEO IDs, the ordinate represents the gene name. Red represents logFC > 0; Green represents logFC < 0; and the value in the box represents the logFC
GO and KEGG pathway enrichment analysis for DEGs
A total of 32 and 173 significantly enriched GO terms (adj. P-value ≤ 0.05) for the up-regulated and down-regulated DEGs, respectively, were identified (Additional file 2: Table S3). The up-regulated genes were mainly enriched in cornification, epithelial cell development, keratinocyte and epidermal cell differentiation, and the ERBB2 signaling pathway (Fig. 2A–B). The down-regulated DEGs were mainly enriched in biological processes related to muscle system and mesenchyme development, extracellular matrix and structure organization, and regulation of cell migration and growth (Fig. 2C–D). KEGG analysis indicated that the integrated DEGs were significantly enriched in seven pathways, including focal adhesion (FA), proteoglycans in cancer, vascular smooth muscle contraction, fluid shear stress and atherosclerosis, leukocyte transendothelial migration, dilated cardiomyopathy (DCM), and tight junctions (Table 2 and Fig. 3A–B).

GO terms analysis for DEGs. A Bubble plot of prior significantly enriched GO terms of up-regulated DEGs in each classification. B GO Chord plot of the relationship between the top eight GO-BP terms and their corresponding up-regulated genes. A gene is linked to a certain GO term by colored bands. C Bubble plot of the prior significantly enriched GO terms of down-regulated DEGs. D GO Chord plot of the relationship between the eight tumor-related enriched GO-BP terms and their corresponding down-regulated genes

KEGG pathway enrichment analysis for DEGs. A Bubble plot of dysregulated pathways. B KEGG Chord plot of the relationship between the selected pathways and their corresponding genes
Multivariate cox analysis and ROC curve plotting for DEGs
According to the AIC, the optimal risk model containing nine genes was determined based on bladder cancer patient gene expression data provided by the TCGA consortium (Table 3). Of these genes, FASN, SPAG4, FCRLB, and UPK2 showed positive coefficients, indicating their role as risk factors for predicting a poor survival. However, UBE2C, FER1L4, EPN3, CTSE, and TOX3 showed negative coefficients, suggesting that they might be protective factors associated with a longer survival. The calculated median value for the risk score of each sample was 1.009. Each sample was grouped into “high-risk” and “low-risk” according to the median risk score. Survival analysis (Fig. 4A) revealed a five-year survival rate of 27.5% [95% confidence interval (CI), 19.3–39.0%] in the high-risk group (202 patients) and 58.1% (95% CI, 49.1–68.7%) in the low-risk group (203 patients). We plotted the ROC curve for the risk model, which yielded an AUC value of 0.726 (Fig. 4B).

Survival and ROC curve analysis for DEGs. A Survival curve. Patients were divided into a high-risk group (> 1.009) and a low-risk group (< 1.009) according to the risk score. The difference in the survival rate between the high-risk and low-risk groups was significant (P < 0.001). B ROC curve. The calculated AUC was 0.726. The larger the AUC value, the more likely the current classification algorithm placed the positive sample in front of the negative sample
Integration of PPI network and module analysis
A PPI network of the integrated DEGs was constructed that comprised 171 nodes and 561 edges (Fig. 5A). Then, the top 30 DEGs ranked by degree value in the PPI network were identified (Fig. 5B). After the module analysis of the PPI network, the top three significant modules were determined (Fig. 5C–E) and enriched in several KEGG pathways. Thirteen genes in module 1 were significantly enriched in vascular smooth muscle and cardiac contraction and pathways associated with cardiomyocyte structural and functional abnormalities. Fourteen genes in module 2 were significantly enriched in proteoglycans in cancer, malaria, and ECM–receptor interaction, and seven genes in module 3 were significantly enriched for HTLV-I infection, cell cycle, and ubiquitin-mediated proteolysis (Additional file 2: Table S4).

PPI network construction and module analysis for DEGs. A The PPI network of DEGs was constructed, and DEGs with ≤ 3 nodes and ≤ 3 edges were removed from the network. Up-regulated genes are marked in red, and down-regulated genes are marked in green. Larger sizes or darker colors of a node indicate that the corresponding gene had a greater logFC. B Bar plot of the nodes with top 30 degrees in the PPI network. Higher numbers and longer bar lengths represent greater interactions of the protein. C The most significant module in the PPI network had 13 nodes and 77 edges. D The second most significant module in the PPI network had 14 nodes and 36 edges. E The third most significant module in the PPI network had 7 nodes and 13 edges
Identification and survival analysis for hub genes
According to the degree value in the PPI network, up-regulated DEGs exhibiting the top five high degree genes (CDH1, IGFBP3, PPARG, SDC1, and EPCAM) and down-regulated DEGs exhibiting the top five high degree genes (ACTA2, COL3A1, TPM1, ACTC1, and ACTN1) were screened out as hub genes. The gene descriptions, fold changes, and corresponding degree values for the hub gene are presented in Table 4. GO analysis for the hub genes identified eleven significantly enriched GO terms, which were the most associated with the differentiation and development of muscle cells and the cytoskeleton (adj. P-value ≤ 0.05) (Additional file 2: Table S5). The hub genes were also confirmed to be expressed in many BC tissue samples (Fig. 6A), showing that CDH1, IGFBP3, and EPCAM were significantly up-regulated in BC samples from different stages, while ACTA2, TPM1, ACTC1, and ACTN1 were significantly down-regulated. In addition, an analysis of the correlation between the OS and DFS of patients with BC and hub genes showed that patients with BC showing altered expression of ACTA2, COL3A1, TPM1, ACTC1, and ACTN1 exhibited a worse OS, while those with altered expression of PPARG had a better OS, and only high-expressed ACTC1 could predict a worse DFS (Fig. 6B).

Expression verification and survival analysis for hub genes. A mRNA expression levels of the ten hub genes using GEPIA online tool. *P < 0.01. B Overall survival (OS) and disease free survival (DFS) analyses and for the hub genes using the GEPIA online tool. Only the genes whose altered expression significantly affect OS and DFS are shown
Correlation between CDH1 expression and clinicopathological characteristics in BC
As the gene with the highest degree in the PPI network, CDH1 was significantly overexpressed in multiple tumor tissues, especially in tumors originating from urogenital tracts, breast, lung, cholangio, cervix, and endometrium, based on TCGA database. However, in colon adenocarcinoma, renal clear cell and papillary cell carcinoma, and thyroid carcinoma, CDH1 was significantly decreased (Fig. 7A). Moreover, CDH1 showed a significantly higher expression in patients with low and high histological grades than in normal BC tissues (Fig. 7B). However, patients with different histological grades, i.e., T, N, and M stages, shared similar CDH1 expression levels (Fig. 7C). Given the results of CDH1 expression in patients with Ta/T1 stage BC, we speculate that CDH1 is markedly up-regulated in the tumor initiation stage but not further altered during the tumor development phase.

Association of CDH1 expression with clinicopathological characteristics and CDH1 methylation with prognosis in BC. A Comparison of CDH1 expression between tumor and para-carcinoma tissues in different types of cancers based on TCGA database. ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001. B CDH1 expression in different histological grade BC tissues and normal bladder tissues. C Patients with different T, N, and M stages shared similar CDH1 expression levels in BC. D Correlation between CDH1 methylation and its expression level in BC. E CDH1 methylation was correlated with prognosis in BC from the MethSurv database. F Heat maps of the association between the methylation level of CDH1 and patient characteristics and genomic subregions
Correlation between methylation of CDH1 and prognosis in BC
Although altered expression of CDH1 did not significantly influence the OS rate in patients with BC, DNA methylation also affects clinical outcomes. The cBioPortal platform was used to evaluate CDH1 methylation in BC, showing that CDH1 expression was highly negatively correlated with methylation (R = − 0.47, P < 0.001) in BC (Fig. 7D). Kaplan–Meier plots by MethSurv analysis were drawn to identify methylation sites in CDH1 associated with prognosis in BC. We found six methylated sites indicating good prognosis (5′-UTR;1stExon-Island-cg11255163 and cg23989635, Body-Open_Sea-cg09220040 and cg07762788, Body-S_Shore-cg09406989, and Body-S_Shelf-cg08616585) and one site indicating an aberrant prognosis (Body-Open_Sea-cg10313337) (Fig. 7E). The heat map plotted by ‘Gene Visualization’ further illustrates the relationship of CDH1 methylation levels with gene subregions and available characteristics of patients (Fig. 7F).
CDH1 expression and identification of correlated genes in BC cell lines
Based on the CCLE database, CDH1 was also significantly overexpressed in several different cancer cells (Fig. 8A), derived from the urinary tract, breast, lung, cholangio, prostate, esophagus, colorectal, and endometrium, which generally conformed to that in tumor tissues. Through co-expression analysis in 25 different urinary tract cancer cell lines extracted from the CCLE database, a total of 755 and 197 genes were identified that were positively and negatively co-expressed with CDH1, respectively (Additional file 2: Table S6). The top 20 positively and negatively co-expressed genes are depicted in a heat map (Fig. 8B). Notably, another hub gene, EPCAM, had a significant positive correlation of 0.805 with CDH1 (Fig. 8C). Both CDH1 and EPCAM were confirmed by qRT-PCR to be up-regulated in 5637 and RT4 BC cells compared to those in the normal urinary tract epithelial cell line SVHUC1 (Fig. 8D). High and low expression CDH1 phenotypes of significantly enriched pathways, including representative metabolic signaling, cell cycle, and RNA degradation pathways (Fig. 8E and Additional file 2: Table S7), were determined using GSEA.

CDH1 expression in vitro and corresponding gene identification and function analysis. A CDH1 expression in various cancer cell lines based on the CCLE database. The ordinate represents the expression level of CDH1. B Heat map of the top 20 positively and negatively co-expressed genes with CDH1 in urinary tract cancer cell lines extracted from the CCLE database. C Correlation between the expression of CDH1 and EPCAM. Cor is the correlation coefficient. D mRNA expression levels of CDH1 and EPCAM in SVHUC1 cells, and 5637 and RT4 bladder tumor cells examined using RT-PCR. Three independent experiments were conducted for each RT-PCR assay, and P < 0.05 was considered to reflect a statistically significant difference from the SVHUC1 group. E GSEA function enrichment analysis of differentially expressed genes in the high CDH1 expression group and low CDH1 expression group based on the CCLE database
Correlation between CDH1 expression and immune cell infiltration in BC
By the ssGSEA method, we quantified the infiltration levels of 24 immune cell types for 413 BC samples of TCGA-BLCA and investigated the association between CDH1 expression and immune cell infiltration. Spearman correlation analyses revealed that high CDH1 expression was mainly associated with low infiltration of the most immune cell types (Fig. 9A), especially pDCs (R = − 0.369, P < 0.001), cytotoxic cells (R = − 0.309, P < 0.001) and Th1 cells (R = − 0.291, P < 0.001). Besides, CD8 + T cells (R = − 0.258, P < 0.001), Treg (R =− 0.233, P < 0.001), T cells (R = − 0.233, P < 0.001), macrophages (R = − 0.216, P < 0.001), neutrophils (R = -0.181, P < 0.001), DCs (R = − 0.175, P = 0.001), B cells (R = − 0.165, P < 0.001), and NK cells (R = − 0.155, P = 0.002) were all negative correlated with CDH1 expression. We observed weakly positive correlations of CDH1 expression level with infiltration of only two immune cell types, including T helper cells (R = 0.1, P = 0.042) and Tcm cells (R = 0.099, P = 0.045). The immune cells relevant to infiltration levels were further evaluated in distinct CDH1 groups (Fig. 9B), which conformed to the results in Fig. 9A.

Correlation between CDH1 expression and immune cell infiltration in BC and the ceRNA network construction. A Relationships among infiltration levels of 24 immune cell types and CDH1 expression profiles in BC samples based on the TCGA database by Spearman correlation analysis. B The infiltration levels of the 24 immune cell types were evaluated in distinct CDH1 groups in BC samples. ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001. DCs, dendritic cells; aDCs, activated DCs; iDCs, immature DCs; pDCs, plasmacytoid DCs; NK, natural killer cells; Th, T helper cells; Th1, type 1 Th cells; Th2, type 2 Th cells; Th17, type 17 Th cells; Tcm, T central memory cells; Tem, T effector memory cells; Tfh, T follicular helper cells; Tgd, T gamma delta cells; Treg, regulatory T cells. C ceRNA network containing hub genes, differently expressed miRNAs and lncRNAs in BC. D ceRNA network associated with CDH1. Larger circular nodes represent mRNA, smaller circular nodes represent miRNA, and hexagonal nodes represent lncRNA
Construction of ceRNA network
We identified 556,431 significantly up-regulated and 125 significantly down-regulated DElncRNAs and 289,181 significantly up-regulated and 108 significantly down-regulated DEmiRNAs between BC and adjacent non-cancer bladder tissues in TCGA database (Additional file 1: Fig S2). Furthermore, a total of 552 miRNAs related to hub genes were predicted by integrating the results obtained in the TargetScan and miRDB databases (Additional file 2: Table S8). Subsequently, a ceRNA network was constructed using DEmiRNAs that interacted with both hub genes and DElncRNAs (Additional file 2: Table S9), including 46 lncRNA nodes, 42 miRNA nodes, and 10 mRNA nodes (Fig. 9C). Of these, hsa-miR-383 was the only significantly down-regulated miRNA interacting with CDH1 in BC and associated with LINC00337, AL356608.1, AL357153.2, LINC00485, MALAT1, and HULC, which were all significantly up-regulated in BC (Fig. 9D).
Discussion
In this study we included four independent datasets of early-stage BC and identified 213 DEGs through integrated analysis. Most GO terms these DEGs enriched were closely associated with the development of BC, such as keratinocyte and epidermal cell differentiation, cell–cell junction, muscle system processes, regulation of cell migration and growth, etc. Besides, DEGs participated in the control of various pathways, including focal adhesion, proteoglycans in cancer, vascular smooth muscle contraction, leukocyte transendothelial migration, and tight junction signaling, contributing to BC progression. FA-related structural molecules are associated with cancer progression and metastasis by promoting cell invasion and epithelial–mesenchymal transition (EMT) [7, 30]. Moreover, emerging evidence supports a critical role of proteoglycans in maintaining homeostasis and carcinogenesis [31]. The contraction of vascular smooth muscle tissue is critical for evaluating vasoactivity [32], and the disruption of tight junctions is a vital step during EMT [33]. Thus, these pathways are potential targets for developing therapeutic strategies for cancer treatment [33, 34]. Leukocyte transendothelial migration has been linked to adhesion molecules and chemokines [35] and is a key step in cancer progression [36]. In this study, an optimal risk model with an AUC value of 0.726 was generated using multivariate Cox analysis of all DEGs, indicating a highly predictive model for patient prognosis of survival in BC.
Normally, the degree of connectivity of a gene in a PPI network reflects its association with corresponding disease, so up-regulated DEGs exhibiting the top five high degree genes (CDH1, IGFBP3, PPARG, SDC1, and EPCAM) and down-regulated DEGs exhibiting the top five high degree genes (ACTA2, COL3A1, TPM1, ACTC1, and ACTN1) were screened out as hub genes. These hub genes were predominantly enriched in pathways associated with muscle cell differentiation and development, regulation of cytoskeleton, and growth factor binding. We compared additional BC and adjacent normal tissues on the GEPIA platform to assess whether the expression trends of these hubs were similar when including intermediate and advanced stage BC tissues as compared to when only early-stage tumors were included. We found that the trends in the expression of CDH1, IGFBP3, EPCAM, ACTA2, TPM1, ACTC1, and ACTN1 in BC samples containing early-advanced stages were consistent with those in early-stage BC. However, significant changes of COL3A1, PPARG, and SDC1 were not detected, implying that their expression appeared to be specifically altered in early-stage BC only. In addition, survival analysis indicated that changes in the expression of ACTA2, COL3A1, TPM1, ACTC1, and ACTN1 were associated with a poor prognosis of BC, while PPARG overexpression indicated a better OS outcome.
Some studies have suggested that PPARG-dependent transcriptional regulation may be involved in the etiology of urothelial carcinoma [37], and decreasing PPARG activity via drug inhibition or gene ablation inhibits the proliferation of BC cells [38]. SDC1 encodes essential cell surface adhesion molecules that maintain cell morphology and a stable microenvironment and promotes tumor progression by stimulating cell proliferation, metastasis, invasion, and angiogenesis [39]. ACTB2 is closely associated with cell motility, structure, and integrity; abnormal expression of this gene accelerates the invasion and metastasis of lung adenocarcinoma [40], suggesting that ACTB2 is a potential mass spectrometry-based diagnostic protein marker for BC [41]. Down-regulation of TPM1 has been detected in both gastric [42] and colorectal cancer [43] during tumor invasion and lymph node metastasis, which are closely associated with BC progression [44], suggesting a role as tumor suppressor. However, increased expression of COL3A1 in BC predicts a poor prognosis [45], consistent with our findings. ACTC1 has been linked to cancer recurrence and OS rate in glioma patients, suggesting that it may be a novel independent marker for prognosis and invasion in glioma [46]. ACTN1 encodes a non-muscular alpha-actin subtype involved in keratinocyte motility by modulating the actin cytoskeleton, focal adhesion, and hemidesmosomal protein complexes, which in turn modulates cell velocity, lipid dynamics, and directional migration [47]. Decreased expression of ACTN1 may improve survival in pancreatic cancer [48]. Recently, IGFBP3 was found to be overexpressed in various tumor types and is not only associated with increased incidence in colorectal cancer [49] but also leads to tumor metastasis by increasing cell migration and adhesion in nasopharyngeal carcinoma [50]. In addition, evidence suggests that genetic polymorphisms in IGFBP3 may be associated with BC tumorigenesis [51]. Brunner et al. [52] found that EPCAM is associated with advanced stage, high grade tumor and poor OS rate in patients with BC, suggesting that it is potential novel predictive marker and a therapeutic target for BC.
CDH1 has the highest degree value in PPI network and was significantly overexpressed in BC tissues, consistent with its overexpression in multiple human cancers. We then found that patients with different histological grades (T, N, and M stages) shared similar CDH1 expression levels. Taking the results of CDH1 expression in Ta/T1 stage BC, we speculate that CDH1 is markedly up-regulated from the initiation tumor stage but not further altered during the tumor development phase, making the value of CDH1 in prediction of BC more prominent than other hub genes. This suggests that high CDH1 expression could predict the presence of early-advanced stage BC, where its expression rises to the peak in the early stage. Notably, although there was no significant correlation between increased CDH1 expression and survival times in our study, loss of CDH1 function contributes to cancer progression by increasing proliferation, invasion, and metastasis in various tumor, such as gastric, breast, and colorectal cancers [53]. Liu Jia et al. reported that in various tumor cells, CDH1 can inhibit PI3K/Akt oncogenic signaling to suppress tumorigenesis [54]. All these findings implied that CDH1 would be a potential therapeutic target for cancers. CDH1 expression was highly negatively correlated with methylation in BC. Methylation of CDH1 is more frequent in BC tissues than in normal control tissues and increasing scientific evidences has suggested that CDH1 gene promoter polymorphism and DNA methylation might contribute to the development and progression of BC [55]. By survival analysis using the MethSurv web tool, patients with high CDH1 methylation generally had a poorer OS rate than patients with low CDH1 methylation. Additionally, several methylated sites in CDH1 associated with prognosis have also been identified, representing abnormal demethylated sites of CDH1 in BC. CDH1 methylation might be a promising prognostic biomarker for BC.
We also investigated the underlying relationship between CDH1 expression and immune cell infiltration in BC. A high CDH1 expression was negatively correlated with immune cell infiltration, such as pDCs, Treg, T cells, macrophages, neutrophils, DCs, and NK cells. Among them, pDCs were the most significantly negatively associated immune cells with a high CDH1 expression. The role of pDCs in different tumor progression stages remains controversial. pDCs can promote tumor cell growth, survival, and drug resistance in multiple myeloma cells and xenograft models [56]. pDC is a unique DC subset, which is considered to play a significant role in immune responses. Activated pDCs can secrete large quantities of type I interferon and are involved in the activation and function regulation of NK cells, B cells, and T cells [57]. pDCs can also induce Treg cell generation, and pDC depletion leads to decreased Treg numbers in the tumor microenvironment [58]. Treg cells have been traditionally regarded as cancer promoters, owing to their function in suppressing antitumor immune responses [59]. In contrast, macrophage and neutrophil infiltration were also negatively correlated with a high CDH1 expression. Different subtypes of macrophages have different effects in tumor development, among which the M2 subtype can promote tumor progression [60]. Tumor neutrophil infiltration also contributes to tumor growth [61]. Therefore, increased CDH1 expression appears to improve tumor immunity by inhibiting pDCs and macrophage and neutrophil accumulation and by reducing Treg generation via the suppression of pDCs to restrict the escape of cancer cells from annihilation and, ultimately, relieve tumorigenesis.
To further explore the mechanism of CDH1 underlying BC tumorigenesis, we investigated the correlated genes of CDH1 in urothelial carcinoma cells and their potentially enriched functions. As a result, another hub gene, EPCAM, exhibited a highly positive correlation with CDH1. We confirmed that both CDH1 and EPCAM expression was up-regulated in BC cells. EPCAM is an epithelial cell adhesion molecule localized on the cell surface and mainly overexpressed in various epithelial malignancies [62]. Increasing evidence suggests that EPCAM is one of the most highly immunogenic tumor-associated antigens. EPCAM promotes proliferation, metastasis, and invasion of tumor cells, but overexpressed EPCAM is associated with a better prognosis in patients with adenocarcinoma of the lung, breast and gall bladder cancer, and squamous cell carcinoma of the esophagus [62]. The actual contribution of EPCAM to tumorigenesis and its prognostic potential for various cancers remain to be explored, which may be mediated via interaction with self-related signaling and other proteins in the plasma membrane, regulation in cancer stem cells, or DNA methylation.
After analyzing the potential function associated with CDH1-related genes, we found that a decreased CDH1 expression corresponded to a significant enrichment of its co-expression partners in many metabolic pathways, cell cycle, and RNA degradation. The propensity of cancer cells to reprogram their metabolism [63] and the disruption of the cell cycle balance [64] have been recognized as key steps in promoting carcinogenesis. Therefore, targeting these metabolic and cell cycle pathways has been the focus of cancer therapy research. As a key factor involved in cell cycle regulation, CDH1 is essential for cell viability and cell cycle progression and regulates the cell cycle by regulating the Claspin/Chk1 and Rb/E2F1 pathways [65]. Furthermore, the activation of the CDH1–APC axis serves an important function during G1 phase arrest and DNA damage-induced G2 phase arrest [66]. EPCAM has also been described as promoter of cell cycle progression, which up-regulates the proto-oncogenes, C-MYC and cyclin A/E [67]. Moreover, the ceRNA analysis revealed that CDH1 may be regulated by miR-383 in BC. miR-383 is a tumor suppressor that inhibits cell proliferation, metastasis, and EMT in BC via targeting ETS1 [68], which can also suppress cell cycle progression in gastric carcinoma cells through regulating Cyclin E2 expression [69]. However, to date, studies on the association between miR-338 and CDH1 are lacking. Therefore, further research is required to verify the correlation between CDH1 and miR-338 underlying BC carcinogenesis and progression.
There were several limitations in this study. First, the sample size was relatively small, and the genetic data lacked ethnic and geographical diversity, potentially influencing our analysis of gene expression in early-stage BC. Second, as gender, age, and pathological typing were not accounted for in this study, it is likely that some biological information is missing.
Conclusions
In summary, CDH1, IGFBP3, PPARG, SDC1, EPCAM, ACTA2, COL3A1, TPM1, ACTC1, and ACTN1, were identified as vital players in the progression of early-stage BC. CDH1 appeared to be significantly up-regulated from the tumor initiation stage, without further alterations during the tumor development phase. Six methylated sites in CDH1 were identified indicating a good prognosis in BC patients and one site for an aberrant outcome. Besides, a high CDH1 expression was negatively correlated with immune cell infiltration, such as pDCs, Treg, T cells, macrophages, neutrophils, DCs, and NK cells. Moreover, EPCAM was highly positively correlated with CDH1, which was predicted to be directly regulated by miR-383 in BC. These biomarkers could serve as potential predictors and therapeutic targets for early-stage BC, which are required to be future confirmed experimentally.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the GEO repository, which can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE3167 (accessed on 10 August 2018); https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE7476 (accessed on 25 March 2019); https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40355 (accessed on 01 February 2019); https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65635 (accessed on 13 August 2019).
Abbreviations
- BP:
-
Biological processes
- CC:
-
Cell component
- CCLE:
-
Cancer cell line encyclopedia
- ceRNA:
-
Competitive endogenous RNA
- CI:
-
Confidence interval
- DEGs::
-
Differentially expressed genes
- DElncRNAs:
-
Differentially expressed lncRNAs
- DEmRNAs:
-
Differentially expressed microRNAs
- DFS:
-
Disease free survival
- FC:
-
Fold change
- FDR:
-
False-discovery rate
- GEO:
-
Gene expression omnibus
- GEPIA:
-
Gene expression profiling interactive analysis
- GO:
-
Gene ontology
- GSEA:
-
Gene set enrichment analysis
- KEGG::
-
Kyoto encyclopedia of genes and genomes
- MCODE:
-
Molecular complex detection
- MF:
-
Molecular function
- NES:
-
Normalised enrichment score
- OS:
-
Overall survival
- PPI:
-
Protein–protein interaction RCC
- ROC:
-
Receiver operating-characteristic
- STRING:
-
Search tool for the retrieval of interacting genes
- TCGA:
-
The cancer genome atlas
References
Feng RM, Zong YN, Cao SM, Xu RH. Current cancer situation in China: good or bad news from the 2018 Global cancer statistics? Cancer Commun (Lond). 2019;39(1):22. https://doi.org/10.1186/s40880-019-0368-6.
Burger M, Catto JW, Dalbagni G, et al. Epidemiology and risk factors of urothelial bladder cancer. Eur Urol. 2013;63(2):234–41. https://doi.org/10.1016/j.eururo.2012.07.033.
Comperat E, Larre S, Roupret M, et al. Clinicopathological characteristics of urothelial bladder cancer in patients less than 40 years old. Virchows Arch. 2015;466(5):589–94. https://doi.org/10.1007/s00428-015-1739-2.
Mostafid H, Babjuk M, Bochner B, et al. Transurethral resection of bladder tumour: the neglected procedure in the technology race in bladder cancer. Eur Urol. 2020;77(6):669–70. https://doi.org/10.1016/j.eururo.2020.03.005.
Crijnen J, De Reijke TM. Emerging intravesical drugs for the treatment of non muscle-invasive bladder cancer. Expert Opin Emerg Drugs. 2018;23(2):135–47. https://doi.org/10.1080/14728214.2018.1474201.
Gakis G. Management of muscle-invasive bladder cancer in the 2020s: challenges and perspectives. Eur Urol Focus. 2020;6(4):632–8. https://doi.org/10.1016/j.euf.2020.01.007.
Gao X, Chen Y, Chen M, et al. Identification of key candidate genes and biological pathways in bladder cancer. PeerJ. 2018;6: e6036. https://doi.org/10.7717/peerj.6036.
Kulasingam V, Diamandis EP. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat Clin Pract Oncol. 2008;5(10):588–99. https://doi.org/10.1038/ncponc1187.
Ni M, Liu X, Wu J, et al. Identification of candidate biomarkers correlated with the pathogenesis and prognosis of non-small cell lung cancer via integrated bioinformatics analysis. Front Genet. 2018;9:469. https://doi.org/10.3389/fgene.2018.00469.
Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10.
Dyrskjot L, Kruhoffer M, Thykjaer T, et al. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004;64(11):4040–8. https://doi.org/10.1158/0008-5472.CAN-03-3620.
Mengual L, Burset M, Ars E, et al. DNA microarray expression profiling of bladder cancer allows identification of noninvasive diagnostic markers. J Urol. 2009;182(2):741–8. https://doi.org/10.1016/j.juro.2009.03.084.
Hecker N, Stephan C, Mollenkopf HJ, et al. A new algorithm for integrated analysis of miRNA-mRNA interactions based on individual classification reveals insights into bladder cancer. PLoS ONE. 2013;8(5): e64543. https://doi.org/10.1371/journal.pone.0064543.
Borisov N, Tkachev V, Suntsova M, et al. A method of gene expression data transfer from cell lines to cancer patients for machine-learning prediction of drug efficiency. Cell Cycle. 2018;17(4):486–91. https://doi.org/10.1080/15384101.2017.1417706.
Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–102. https://doi.org/10.1093/nar/gkx247.
Barretina J, Caponigro G, Stransky N, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. https://doi.org/10.1038/nature11003.
Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7): e47. https://doi.org/10.1093/nar/gkv007.
Kolde R, Laur S, Adler P, Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28(4):573–80. https://doi.org/10.1093/bioinformatics/btr709.
Yu G, Wang LG, Yan GR, He QY. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 2015;31(4):608–9. https://doi.org/10.1093/bioinformatics/btu684.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7. https://doi.org/10.1089/omi.2011.0118.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. https://doi.org/10.1093/nar/28.1.27.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51. https://doi.org/10.1002/pro.3715.
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49(D1):D545–51. https://doi.org/10.1093/nar/gkaa970.
Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. https://doi.org/10.1073/pnas.0506580102.
Franceschini A, Szklarczyk D, Frankild S, et al. STRING v.91: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:808–15.
Bandettini WP, Kellman P, Mancini C, et al. Multicontrast delayed enhancement (MCODE) improves detection of subendocardial myocardial infarction by late gadolinium enhancement cardiovascular magnetic resonance: a clinical validation study. J Cardiovasc Magn Reson. 2012;14:83. https://doi.org/10.1186/1532-429X-14-83.
Modhukur V, Iljasenko T, Metsalu T, et al. MethSurv: a web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics. 2018;10(3):277–88. https://doi.org/10.2217/epi-2017-0118.
Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. https://doi.org/10.1186/1471-2105-14-7.
Aho K, Derryberry D, Peterson T. Model selection for ecologists: the worldviews of AIC and BIC. Ecology. 2014;95(3):631–6. https://doi.org/10.1890/13-1452.1.
Yamasaki T, Seki N, Yoshino H, et al. MicroRNA-218 inhibits cell migration and invasion in renal cell carcinoma through targeting caveolin-2 involved in focal adhesion pathway. J Urol. 2013;190(3):1059–68. https://doi.org/10.1016/j.juro.2013.02.089.
Peng Q, Shen Y, Lin K, et al. Identification of microRNA-92a and the related combination biomarkers as promising substrates in predicting risk, recurrence and poor survival of colorectal cancer. J Cancer. 2019;10(14):3154–71. https://doi.org/10.7150/jca.30306.
Tseng H, Gage JA, Haisler WL, et al. A high-throughput in vitro ring assay for vasoactivity using magnetic 3D bioprinting. Sci Rep. 2016;6:30640. https://doi.org/10.1038/srep30640.
Kyuno D, Yamaguchi H, Ito T, et al. Targeting tight junctions during epithelial to mesenchymal transition in human pancreatic cancer. World J Gastroenterol. 2014;20(31):10813–24. https://doi.org/10.3748/wjg.v20.i31.10813.
McCrudden CM, O’Rourke MG, Cherry KE, et al. Vasoactivity of rucaparib, a PARP-1 inhibitor, is a complex process that involves myosin light chain kinase, P2 receptors, and PARP itself. PLoS ONE. 2015;10(2): e0118187. https://doi.org/10.1371/journal.pone.0118187.
Nhu TQ, Bich Hang BT, Cornet V, et al. Single or Combined dietary supply of psidium guajava and phyllanthus amarus extracts differentially modulate immune responses and liver proteome in striped catfish (Pangasianodon hyphophthalmus). Front Immunol. 2020;11:797. https://doi.org/10.3389/fimmu.2020.00797.
Camaj PR, Graziano JH, Preteni E, et al. Long-term effects of environmental lead exposure on blood pressure and plasma soluble cell adhesion molecules in young adults: a follow-up study of a prospective cohort in Kosovo. J Environ Public Health. 2018;2018:3180487. https://doi.org/10.1155/2018/3180487.
Liu C, Tate T, Batourina E, et al. Pparg promotes differentiation and regulates mitochondrial gene expression in bladder epithelial cells. Nat Commun. 2019;10(1):4589. https://doi.org/10.1038/s41467-019-12332-0.
Goldstein JT, Berger AC, Shih J, et al. Genomic activation of PPARG reveals a candidate therapeutic axis in bladder cancer. Cancer Res. 2017;77(24):6987–98. https://doi.org/10.1158/0008-5472.CAN-17-1701.
Akl MR, Nagpal P, Ayoub NM, et al. Molecular and clinical profiles of syndecan-1 in solid and hematological cancer for prognosis and precision medicine. Oncotarget. 2015;6(30):28693–715.
Lee HW, Park YM, Lee SJ, et al. Alpha-smooth muscle actin (ACTA2) is required for metastatic potential of human lung adenocarcinoma. Clin Cancer Res. 2013;19(21):5879–89. https://doi.org/10.1158/1078-0432.CCR-13-1181.
Wu Z, Wang S, Jiang F, et al. Mass spectrometric detection combined with bioinformatic analysis identified possible protein markers and key pathways associated with bladder cancer. Gene. 2017;626:407–13. https://doi.org/10.1016/j.gene.2017.05.054.
Hu L, Fang L, Zhang ZP, Yan ZL. TPM1 is a novel predictive biomarker for gastric cancer diagnosis and prognosis. Clin Lab. 2020. https://doi.org/10.7754/Clin.Lab.2019.190235.
Ge T, Xiang P, Mao H, et al. Inhibition of miR-96 enhances the sensitivity of colorectal cancer cells to oxaliplatin by targeting TPM1. Exp Ther Med. 2020;20(3):2134–40. https://doi.org/10.3892/etm.2020.8936.
Liu G, Zhao X, Zhou J, et al. Long non-coding RNA MEG3 suppresses the development of bladder urothelial carcinoma by regulating miR-96 and TPM1. Cancer Biol Ther. 2018;19(11):1039–56. https://doi.org/10.1080/15384047.2018.1480279.
Yuan L, Shu B, Chen L, et al. Overexpression of COL3A1 confers a poor prognosis in human bladder cancer identified by co-expression analysis. Oncotarget. 2017;8(41):70508–70520 https://doi.org/10.18632/oncotarget.19733.
Ohtaki S, Wanibuchi M, Kataoka-Sasaki Y, et al. ACTC1 as an invasion and prognosis marker in glioma. J Neurosurg. 2017;126(2):467–75. https://doi.org/10.3171/2016.1.JNS152075.
Hamill KJ, Hiroyasu S, Colburn ZT, et al. Alpha actinin-1 regulates cell-matrix adhesion organization in keratinocytes: consequences for skin cell motility. J Invest Dermatol. 2015;135(4):1043–52. https://doi.org/10.1038/jid.2014.505.
Rajamani D, Bhasin MK. Identification of key regulators of pancreatic cancer progression through multidimensional systems-level analysis. Genome Med. 2016;8(1):38. https://doi.org/10.1186/s13073-016-0282-3.
Chan YX, Alfonso H, Paul Chubb SA, et al. Higher IGFBP3 is associated with increased incidence of colorectal cancer in older men independently of IGF1. Clin Endocrinol (Oxf). 2018;88(2):333–40. https://doi.org/10.1111/cen.13499.
Bao L, Liu H, You B, et al. Overexpression of IGFBP3 is associated with poor prognosis and tumor metastasis in nasopharyngeal carcinoma. Tumour Biol. 2016;37(11):15043–52. https://doi.org/10.1007/s13277-016-5400-8.
Safarinejad MR, Shafiei N, Safarinejad SH. The association between bladder cancer and a single nucleotide polymorphism (rs2854744) in the insulin-like growth factor (IGF)-binding protein-3 (IGFBP-3) gene. Arch Toxicol. 2011;85(10):1209–18. https://doi.org/10.1007/s00204-011-0671-8.
Brunner A, Prelog M, Verdorfer I, et al. EpCAM is predominantly expressed in high grade and advanced stage urothelial carcinoma of the bladder. J Clin Pathol. 2008;61(3):307–10. https://doi.org/10.1136/jcp.2007.049460.
Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat Struct Mol Biol. 2009;16(9):979–86. https://doi.org/10.1038/nsmb.1663.
Liu J, Wan L, Liu J, et al. Cdh1 inhibits WWP2-mediated ubiquitination of PTEN to suppress tumorigenesis in an APC-independent manner. Cell Discov. 2016;2:15044. https://doi.org/10.1038/celldisc.2015.44.
Wang Y, Kong CZ, Zhang Z, Yang CM, Li J. Role of CDH1 promoter polymorphism and DNA methylation in bladder carcinogenesis: a meta-analysis. DNA Cell Biol. 2014;33(4):205–16. https://doi.org/10.1089/dna.2013.2100.
Chauhan D, Singh AV, Brahmandam M, et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: a therapeutic target. Cancer Cell. 2009;16(4):309–23. https://doi.org/10.1016/j.ccr.2009.08.019.
Chen Y, Yang JE, Tang JM, et al. Predictive value of plasmacytoid dendritic cells and Toll-like receptor-9 regarding the treatment efficacy of interferon-alpha in HBeAg-positive chronic hepatitis B patients. Exp Ther Med. 2019;18(6):4541–6. https://doi.org/10.3892/etm.2019.8161.
Sawant A, Hensel JA, Chanda D, et al. Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells. J Immunol. 2012;189(9):4258–65. https://doi.org/10.4049/jimmunol.1101855.
Dennis KL, Blatner NR, Gounari F, Khazaie K. Current status of interleukin-10 and regulatory T-cells in cancer. Curr Opin Oncol. 2013;25(6):637–45. https://doi.org/10.1097/CCO.0000000000000006.
Huang R, Liu J, Li H, et al. Identification of hub genes and their correlation with immune infiltration cells in hepatocellular carcinoma based on GEO and TCGA databases. Front Genet. 2021;12: 647353. https://doi.org/10.3389/fgene.2021.647353.
Wang Y, Wang K, Han GC, et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol. 2014;7(5):1106–15. https://doi.org/10.1038/mi.2013.126.
Baeuerle PA, Gires O. EpCAM (CD326) finding its role in cancer. Br J Cancer. 2007;96(3):417–23. https://doi.org/10.1038/sj.bjc.6603494.
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14. https://doi.org/10.1126/science.123.3191.309.
Ma X, Edmonson M, Yergeau D, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun. 2015;6:6604. https://doi.org/10.1038/ncomms7604.
Gao D, Inuzuka H, Korenjak M, et al. Cdh1 regulates cell cycle through modulating the claspin/Chk1 and the Rb/E2F1 pathways. Mol Biol Cell. 2009;20(14):3305–16. https://doi.org/10.1091/mbc.E09-01-0092.
Sudo T, Ota Y, Kotani S, et al. Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells. EMBO J. 2001;20(22):6499–508. https://doi.org/10.1093/emboj/20.22.6499.
Munz M, Kieu C, Mack B, et al. The carcinoma-associated antigen EpCAM upregulates c-myc and induces cell proliferation. Oncogene. 2004;23(34):5748–58. https://doi.org/10.1038/sj.onc.1207610.
Zhang L, Yan R, Zhang SN, et al. MicroRNA-338–3p inhibits the progression of bladder cancer through regulating ETS1 expression. Eur Rev Med Pharmacol Sci. 2019;23(5):1986–95. https://doi.org/10.26355/eurrev_201903_17237.
Zhu C, Huang Q, Zhu H. miR-383 inhibited the cell cycle progression of gastric cancer cells via targeting cyclin E2. DNA Cell Biol. 2019;38(8):849–56. https://doi.org/10.1089/dna.2019.4624.
Acknowledgements
We appreciate the public database provider and maintenance staffs.
Funding
This work is supported by Jiangsu Medical Innovation Team (CXTDA-2017–48); Key Projects of Jiangsu Science and Technology Plan (BE2019637, BE2020758); High-level health talents "Six One Project" top talents (LGY2019058); the Key Project of Xuzhou Science and Technology (KC19075 and KC18036); Xuzhou Medical Outstanding Talents (YXJCRC-2017–02); the Backbone of Clinical technical training program of Xuzhou (2020GG019); Young Medicine and Technology Innovation Project of Xuzhou (XWKYSL20210277).
Author information
Authors and Affiliations
Contributions
All authors participated in the design, interpretation of the studies and review of the manuscript. TF, LX, YD and HH performed data acquisition and statistical analyses. LH, WZ, WM, BD, GZ and CH provided technical support. TF, LX and HH drafted the manuscript. TF, BD, CH, YD and HH provided critical comments, suggestions, and revised the manuscript. LH, CH and YD provided funding support. All authors read and approved by the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Normalisation of gene expression. Figure S2. DElncRNAs and DEmiRNAs in patients with BC.
Additional file 2: Table S1.
The clinical information of the samples in each Dataset. Table S2. Information of the integrated DEGs. Table S3. GO terms enrichment analysis of EGs. Table S4. KEGG enrichment for the top significant modules of the PPI network. Table S5. GO terms analysis for the hub genes. Table S6. Genes correlated with CDH1 based on CCLE database. Table S7. The GSEA results for CDH1 in BC cell lines. Table S8. The miRNAs related to hub genes. Table S9. The miRNAs paired with DElncRNAs and hub genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fan, T., Xue, L., Dong, B. et al. CDH1 overexpression predicts bladder cancer from early stage and inversely correlates with immune infiltration. BMC Urol 22, 156 (2022). https://doi.org/10.1186/s12894-022-01103-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12894-022-01103-7