
Abstract
Background
Protein kinase C (PKC) is expressed in many tissues and organs including the urinary bladder, however, its role in bladder physiology and pathophysiology is still evolving. The aim of this review was to evaluate available evidence on the involvement of PKC in regulation of detrusor contractility, muscle tone of the bladder wall, spontaneous contractile activity and bladder function under physiological and pathophysiological conditions.
Methods
This is a non-systematic review of the published literature which summarizes the available animal and human data on the role of PKC signaling in the urinary bladder under different physiological and pathophysiological conditions. A wide PubMed search was performed including the combination of the following keywords: “urinary bladder”, “PKC”, “detrusor contractility”, “bladder smooth muscle”, “detrusor relaxation”, “peak force”, “detrusor underactivity”, “partial bladder outlet obstruction”, “voltage-gated channels”, “bladder nerves”, “PKC inhibitors”, “PKC activators”. Retrieved articles were individually screened for the relevance to the topic of this review with 91 citations being selected and included in the data analysis.
Discussion
Urinary bladder function includes the ability to store urine at low intravesical pressure followed by a subsequent release of bladder contents due to a rapid phasic contraction that is maintained long enough to ensure complete emptying. This review summarizes the current concepts regarding the potential contribution of PKC to contractility, physiological voiding, and related signaling mechanisms involved in the control of both the storage and emptying phases of the micturition cycle, and in dysfunctional voiding. Previous studies linked PKC activation exclusively with an increase in generation of the peak force of smooth muscle contraction, and maximum force generation in the lower urinary tract. More recent data suggests that PKC presents a broader range of effects on urinary bladder function including regulation of storage, emptying, excitability of the detrusor, and bladder innervation.
Summary
In this review, we evaluated the mechanisms of peripheral and local regulation of PKC signaling in the urinary bladder, and their impact on different phases of the micturition cycle under physiological and pathophysiological conditions.
Similar content being viewed by others

Background
Physiological contractions of the detrusor smooth muscle (DSM) involve the ability to generate and maintain contractile force during the emptying phase of the micturition cycle [1–13]. Initial animal studies suggested that PKC plays a minimal role in DSM contractility [14–19]. This conclusion was based on the failure of PKC to make a significant contribution to maximal force generation in response to muscarinic receptor agonists [14, 16, 18]. Subsequent investigations provided evidence that PKC likely has dual effects on bladder function regulating both contractility and relaxation of the detrusor [3, 7, 8, 20, 21]. For instance, it was determined that low levels of PKC stimulation could inhibit spontaneous contractions and lower basal resting tone of DSM in vitro, thereby, likely contributing to bladder storage [8], while high levels of PKC activity contributed to an increase and maintenance of total DSM force (integral force) required for bladder emptying [22].
Stimulation of muscarinic receptors triggers activation of phospholipase C (PLC)/PKC downstream signaling associated with smooth muscle contractility via G-protein-coupled receptors [15, 16, 23, 24]. Animal studies confirmed that the same pathway is present in the urinary bladder [15], and that PKC is involved in a variety of cellular events that regulate DSM contractility. Among these are the effects on the smooth muscle itself [3, 7, 8, 17, 25–30], expression and function of ion channels [8, 26, 31–33], and intrinsic bladder nerves assessed by in vitro contractility studies on isolated DSM strips during electric field stimulation (EFS) [22, 34, 35]. In this review, we summarized current data on the involvement of PKC signaling in modulation of bladder function with focus on the contractile force of DSM, relaxation of the muscle during filling phase, as well as the ability of the detrusor to develop and maintain muscle tone throughout the micturition cycle under physiological and pathophysiological conditions.
Methods
This is a non-systematic review of the published literature which summarizes the available animal and human data on the role of PKC signaling in the urinary bladder under different physiological and pathophysiological conditions. A wide PubMed search was performed including the combination of the following keywords: “urinary bladder”, “PKC”, “detrusor contractility”, “bladder smooth muscle”, “detrusor relaxation”, “peak force”, “detrusor underactivity”, “partial bladder outlet obstruction”, “voltage-gated channels”, “bladder nerves”, “PKC inhibitors”, “PKC activators”. Retrieved articles were individually screened for the relevance to the topic of this review with 91 citations being selected and included in the analysis.
Table 1 summarizes the effects of a variety of PKC inhibitors, activators, and cholinergic agonists on PKC signaling and contractility of DSM.
Discussion
The aim of this review was to evaluate the available evidence on the involvement of PKC in regulation of detrusor contractility, muscle tone of the bladder wall, spontaneous contractile activity and bladder function under physiological and pathophysiological conditions.
Modulation of DSM excitability and ion channel activity by PKC
Bladder smooth muscle cells express a variety of ion channels controlling cell excitability. Voltage-gated calcium channels (VGCC), large- (BK), and small- (SK) conductance Ca2+-activated potassium channels, and ATP-sensitive potassium channels (KATP) are the main channels involved in the control of detrusor excitability and excitation-contraction coupling [4, 15, 26, 32, 36].
Voltage gated calcium channels
Several types of VGCC are expressed in the detrusor [15] with L-type VGCC being the main ion channel responsible for the onset of action potentials in DSM cell, as well as for the contractile response of the detrusor to muscarinic receptor stimulation [37–42]. VGCC have also been shown to mediate spontaneous contractions in DSM cells [4]. Recent studies determined that one of the PKC activators, phorbol-12,13-dibutyrate (PDBu), inhibited voltage-dependent spontaneous contractions in isolated DSM strips from rabbit and rat bladders at low concentrations [8, 22]. Additionally, PKC activation by PDBu inhibited receptor-induced phasic activity in the bladders of newborn mice but not in adult bladders [29]. Inhibition of DSM spontaneous contractility by PKC activator, PDBu, was also observed in isolated muscle strips of the human DSM (unpublished observations from our laboratory) and rat DSM [22]. Utilizing whole-cell patch clamp technique, Kajioka et al. (2002) demonstrated an increase in the amplitude and current density of L-type Ca2+ channels upon depolarization in pig and human bladders [43]. Interestingly, in the same study, carbachol significantly diminished the amplitude of VGCC current which was sensitive to nifedipine. The mechanisms of the opposing actions of carbachol on VGCC are not entirely clear. It is well established that initial phasic contraction of DSM in response to carbachol stimulation is associated with membrane depolarization and opening of VGCC [15]. However, this initial massive increase in intracellular calcium is followed by subsequent closing of VGCC and a decrease in intracellular Ca2+ due to its storage in the intracellular stores [44]. These subsequent changes are associated with a quiescent state of the muscle after initial phasic contraction, and, likely, underlie the absence of spontaneous contractile activity during relaxation phase after muscarinic stimulation (Fig. 1). This action may be mediated by carbachol-induced activation of PKC [24] at low or resting calcium concentration, since PKC stimulation by PDBu was reported to activate BK channels resulting in inhibition of spontaneous contractions [8, 22]. Interestingly, pre-incubation of the DSM strips in vitro with the PKC inhibitor, Bim-1, before carbachol stimulation preserved spontaneous contractility during relaxation phase (Fig. 1).

Effects of carbachol (CCh) on the contractile force and spontaneous contractions in DSM in vitro. Isolated muscle strips from rabbit bladders were mounted in organ baths with Tyrode’s buffer (equilibrated with 95%O2/5%CO2) and allowed to develop spontaneous contractions. The muscle strip shown in a was treated with 20 μM of carbachol while the muscle strip in b was first pre-incubated with Bim-1 (28 nM), a PKC inhibitor, for 45 min prior to adding carbachol. Treatment with carbachol increased peak muscle force and reduced the amplitude of spontaneous contraction (SCA) by 96 ± 14 %, (n = 4, p < 0.05 in a), however, carbachol had no effect on spontaneous contraction amplitude (SCA) after PKC inhibition with Bim-1 (b). c Normalized amplitude of the peak force of the contractile response to CCh. Maximal amplitude was taken as 100 %. Light bars show the effects of carbachol on peak force and SCA without Bim-1. Dark bars show the effect of carbachol on peak force and SCA with Bim-1 application
Effects of PKC activation on BK channels
The storage phase of the micturition cycle requires a quiescent smooth muscle that can accommodate increasing volumes at low intravesical pressure. Potassium channels contribute to this process by helping maintain the resting membrane potential via regulation of both the intracellular calcium concentration, and calcium entry into the cell from extracellular sources [32, 36, 45]. Several types of potassium channels are expressed in DSM, and are known to be involved in this regulation. Among them, BK and KATP channels were shown to be regulated by PKC. PKC was shown to be involved in the regulation of BK channels expressed in DSM tissue of rabbit [8] and guinea pig [31] bladders. Hypolite et al. (2013) reported that low levels of PKC stimulation by the PKC activator, PDBu, inhibited spontaneous myogenic contractions, and reduced basal DSM tone in rabbit DSM, which was dependent on activation of BK channels [8]. Additionally, PDBu was unable to inhibit spontaneous contractions in the presence of the BK channel blocker, iberiotoxin, suggesting that the mechanism of PDBu-induced inhibition of spontaneous contractions was via activation of BK channels. Utilizing whole-cell patch clamp recordings in guinea pig DSM cells, Hristov et al. (2014) confirmed that PKC does regulate BK channel activity, however, these authors reported that PKC stimulation with PMA increased muscle force along with amplitude of spontaneous contractions [31]. The variability in the responses to PKC stimulation reported by the abovementioned studies could be attributed to different species (guinea pigs vs rabbits), or the use of distinct PKC activators (PDBu versus PMA), as well as slightly different methodological approaches.
Modulation of KATP channels by PKC
Similar to BK channels, KATP channels also participate in the control of detrusor excitability across a broad spectrum of mammalian species including guinea pigs [46, 47], humans [48], and rats [49]. Activation of these channels induces DSM relaxation in response to KATP channel openers pinacidil [50], and cromakalim [51]. Activation of M3 receptors can induce inhibition of these channels in smooth muscle cells via PKC signaling pathways [52]. Stimulation of muscarinic receptors by carbachol was shown to inhibit KATP currents by 60.7 %, while activators of PKC inhibited KATP channels by 74 % [52]. Additionally, PKC blockers used before stimulation with muscarinic receptor agonists, significantly reduced carbachol-induced inhibition of KATP currents confirming that muscarinic-dependent inhibition of KATP currents is mediated via PKC pathways [52]. This speculation is consistent with in vitro studies showing that the PKC inhibitor, Bim-1, reduced both intrinsic basal tone, and maintained force [8], while awake cystometry performed in rats revealed that inhibition of PKC resulted in increased frequency of urination, and decreased void volume [22]. Schematic presentation of the ion channels involved in regulation of BSM excitability and contractility as well as downstream signaling including PKC pathways is depicted in Fig. 2.

Schematic presentation of the PKC signaling pathways involved in the regulation of urinary bladder function. Abbreviations are spelled out in the text of the manuscript. (+) Activation of the pathways, (−) means Inhibition, (P) is Phosphorylation. Broken arrows indicate potential pathways for PKC-induced inhibition of DSM tone via activation of BK channels
The link between muscarinic receptors and PKC signaling in the control of detrusor contractility
Muscarinic receptor type 3 (M3) signaling via Gq/11 G-protein-coupled receptors includes activation of PLC/PKC as determined for several types of smooth muscle [23] including the DSM of the bladder [16, 53–55]. Initial experiments in the urinary bladder established that inhibition of PLC/PKC pathway by a number of specific inhibitors had no significant effect on maximal contractility of rat bladder muscle strips in response to carbachol stimulation leading to the conclusion that PLC/PKC-dependent signaling via M3 receptors does not significantly contribute to DSM contractility [16]. This data was further buttressed by the studies showing that direct inhibition of PKC had no significant effect on the peak contractile force in response to both carbachol and EFS stimulation [8, 14].
An exception to the above findings was the observation that one of the PKC inhibitors, Bim-1, did reduce maximal force in response to carbachol upon application of a higher concentration of the inhibitor [24]. Another PKC inhibitor, GF 109203X (1 μM), was also reported to reduce peak force in control bladders by 29 % [56]. Subsequent studies clarified that inhibition of PKC with Bim-1, at lower concentrations, did not significantly affect electric field stimulation (EFS)-induced maximal amplitude of DSM contractions, however, significantly reduced the integral, or total force [8, 22]. The integral (total) force is calculated as the area under the curve of a single recorded contraction, and represents the ability of DSM to maintain muscle force required for physiological bladder emptying [8]. The effects of Bim-1 observed in vitro were further confirmed in vivo by cystometric recordings in unanesthetized rats [22]. During urodynamic recordings (cystometry), the urinary bladder was slowly infused with a solution containing Bim-1, causing the voided urine volumes to decline progressively in comparison with infusion of saline in the control group. These in vivo results supported the importance of PKC for muscle force maintenance associated with bladder emptying.
Additional studies confirmed the involvement of PKC in the maintenance of muscle force at the molecular and biochemical level [24]. Stimulation of muscarinic receptors in the smooth muscle results in inhibition of myosin light chain (MLC) phosphatase activity, an increase in MLC phosphorylation and, therefore, contractile force. One of the underlying pathways for inhibition of MLC phosphatase activity is protein kinase C (PKC)-catalyzed phosphorylation of CPI-17 protein [57, 58]. Wang and co-authors evaluated phosphorylation of protein complex Thr(38)-CPI-17, the downstream target of PKC signaling, and established that PKC activation increased Thr(38)-CPI-17 phosphorylation throughout the phasic and tonic portions of carbachol stimulation confirming that PKC is involved in maintaining the contractile force in the detrusor [24].
Limited contribution of the PLC/PKC pathway to the peak amplitude of the contractile response in DSM seems to be physiologically justified as the maximal contractile force of smooth muscle is predominantly controlled by calcium/calmodulin-dependent activation of myosin light chain kinase (MLCK) followed by subsequent phosphorylation of the myosin light chain (MLC) [59]. Since stimulation of DSM by muscarinic receptor agonists leads to a rapid rise in intracellular calcium followed by an equally rapid decline of the calcium to almost basal levels [44], it is highly likely that PKC-induced calcium sensitization via Gq11 receptors [3] plays a major role in the maintenance phase of the contraction that is critical for bladder emptying. This mechanism may partly explain why inhibition of PKC significantly reduces DSM force maintenance, and bladder emptying, without significantly affecting peak force generation [14, 22].
Role of PKC in regulation of spontaneous contractions in the urinary bladder
Protein kinase C is also involved in other aspects of DSM contractility, including the inhibition of basal DSM tone, and spontaneous myogenic contractions [8, 22]. For instance, PDBu, a PKC activator, inhibits spontaneous contractions, and basal myogenic tone at low levels of application (1–50 nM) whereas high concentrations of the drug increase the sensitivity to EFS stimulation in vitro, and also trigger micturition contractions in vivo confirming a dual, concentration-dependent activation profile of PKC effects in DSM [22]. The ability of PKC to modulate basal DSM tone, and inhibit spontaneous contractions at low levels of stimulation may have physiological implications for bladder storage function, while its contribution to the maintenance of DSM force and nerve-mediated contractions may be important for bladder emptying.
Spontaneous myogenic contractions of DSM have been recorded in vivo and in vitro in several species including rabbit [8, 11], rat [60], guinea pig [31], pig [40], and humans [61]. The mechanism of these contractions involves calcium entry into the smooth muscle cells via VGCC [40, 43, 62, 63] to trigger action potential generation, while the repolarization phase is thought to be mediated mainly by opening of calcium-activated potassium channels and exit of potassium ions from the smooth muscle cells leading to muscle relaxation [64]. Early reports suggested that spontaneous large-amplitude contractions recorded in vitro in isolated DSM strips were likely an artifact with no significant functional role [11]. Subsequent studies suggested that these contractions participate in the maintenance of an intrinsic DSM tone by helping the smooth muscle adjust its length and shape in response to bladder filling [4]. They have also been reported to be associated with peripheral sensory processing that informs the need to void as the bladder approaches capacity [6].
The inhibitory effect of the PKC activator, PDBu, on spontaneous contractions was linked to activation of BK channels, subsequent hyperpolarization of the membrane, and restriction of calcium entry via VGCC [8, 65]. Freshly isolated DSM strips from the rabbit bladder exhibit high amplitude spontaneous contractions which decline over several hours, however, the amplitude of contractions remains at a high level in the presence of PKC inhibitor Bim-1 [8]. This observation suggests that endogenous PKC signaling likely maintains the amplitude of spontaneous contractions at a low, physiological level under normal physiological conditions. This assumption is supported by the fact that commercially available PKC activator, PDBu, at low levels of stimulation (1–50 nM), accelerated the decline in amplitude of spontaneous contractions over time in vitro [8]. PDBu was also reported to inhibit carbachol-induced phasic contractions in neonatal bladders associated with the effects on T-type channels [29], and was also observed to inhibit spontaneous contractions in rat DSM [22].
Effects of calcium on PKC activity
Protein kinase C conveys both calcium-dependent [66–68], and calcium-independent [3, 68, 69] effects on DSM contractility in vitro mediated via inhibition of MLCP. It is still unknown if these separate effects are mediated by different PKC isoforms. However, the ability of PKC to mediate both of these actions in DSM may be well suited to urinary bladder storage and emptying function. The storage phase requires the maintenance of basal intrinsic DSM tone at low calcium concentrations which can be mediated, in part, by calcium-independent contractile effects of PKC involved in an increase in tone during the storage phase. This process may be aided by the stretch of the bladder wall associated with an up-regulation of the proteins involved in calcium sensitization [70] and contributing to the basal tone during the early phase of the micturition cycle. However, as the bladder continues to expand, it is likely that enhanced stretch-induced calcium release [71], and calcium-dependent PKC activity increase wall tension further as the bladder approaches capacity. This dual, calcium-independent and dependent, flexibility of PKC may be vital in helping ensure that wall tension rises in a controlled fashion so as to prevent steep increases in bladder pressure during the storage phase of the micturition cycle. PKC-dependent effects have been observed in DSM in response to EFS, which activates the intramural nerves, suggesting that PKC-dependent regulation of neuronal function in DSM is also a distinct possibility [65, 72–74].
PKC signaling in the human bladder
In comparison to extensive animal data, very little information is available regarding the effects of PKC on human bladder contractility and relaxation. Early studies using human tissues suggested that PKC did not significantly contribute to human bladder contractions [14]. However, only the peak amplitude of contraction (maximum force) was evaluated in the presence of various PKC inhibitors, and no difference was found in peak force generation in comparison to the absence of inhibitors. Since bladder emptying requires both, the generation of peak force and the ability to maintain force, it is possible that PKC could play a more significant role in force maintenance in the human DSM in addition to force generation.
A recent study [75] provided evidence that carbachol, a muscarinic receptor agonist, could indirectly inhibit large conductance Ca2+-activated potassium (BK) channels in human DSM cells leading to increased excitability [76]. This is an interesting finding as the ongoing studies in our laboratory using human DSM indicate that carbachol can induce a significant increase in phasic contractions in some human DSM isolated strips, but not in all of them (unpublished data). It is possible that PKC could be an important secondary messenger between muscarinic receptors and BK channels in the human detrusor. Therefore, further studies are warranted to comprehensively characterize the role of PKC in the regulation of cholinergic activity in the human DSM under normal and pathologic conditions.
Regulation of bladder function by PKC under pathophysiological conditions
The role of PKC in bladder pathophysiology is largely related to DSM contractile dysfunction and dysfunctional voiding associated with partial bladder outlet obstruction (PBOO), and detrusor overactivity (DO). Initial studies, using a PBOO model in rabbits, reported that PKC signaling under pathophysiological conditions was uncoupled from its downstream targets and produced little or no force in response to PKC activator, PDBu [27]. Chang et al. [25] also determined that decreased force generation in decompensated PBOO bladders in response to PKC activator, PDBu, was associated with reduced expression of PKC and increased frequency of urination in the obstructed bladders. It should be noted, however, that a broad range of contractile and metabolic dysfunctions were linked to decompensated bladder function in PBOO models [77, 78]. Further studies confirmed a deficit in the PKC pathway in guinea pig DSM after PBOO [79]. Partial bladder outlet obstruction causes two phenotypes of bladder dysfunction referred to as compensated and decompensated bladders [25, 77]. Compensated bladders are characterized mainly by increased smooth muscle hypertrophy, however, residual urine and frequency of micturition have been reported in compensated bladders associated with PKC dysfunction [25, 80, 81]. Decompensated bladders develop an increase in extracellular matrix including collagen and connective tissue resulting in a loss of bladder compliance, decreased contractility, and a substantial loss of bladder emptying function [25, 77, 78, 80–83]. Changes in PKC expression and activity in a PBOO model seem to be dependent on the species and degree of obstruction. Thus, in the rabbit model of PBOO, decompensated bladders revealed a reduction in PKC expression, activity and force generation associated with reduced bladder emptying and frequency of urination, while compensated bladders exhibited increased PKC expression [25]. However, in the rat bladders with PBOO, PKC expression was reduced in both compensated, and decompensated bladders [81]. Additionally, recent in vivo studies established that inhibition of PKC during cystometric recordings in awake rats induced bladder decompensation characterized by an increase in non-voiding contractions (DO), and a significant decrease in void volume [22].
Increased PKC-mediated frequency of urination along with DO have also been reported in the absence of PBOO. In a model of obesity associated with insulin resistance, PKC expression was significantly higher in the bladders of obese mice [26] which showed increased contractility to PDBu, increased frequency, and non-voiding contractions. The changes in the voiding cycle were reversed by metformin treatment which restored high PKC expression in the urinary bladders. High levels of PKC stimulation in rat bladders [22], and high level of expression in compensated rabbit bladders [25] were associated with enhanced nerve-mediated contractions, and frequency of urination, respectively. Thus, a high level of PKC activity and expression may lower the threshold for activation of intramural nerves leading to neurogenic frequency of urination in both diabetic, and compensated bladders. It was established that the protein adiponectin, expressed in adipose tissue, contributed to the increased expression of calcium-dependent PKC-α and enhanced contractile force of DSM in adiponectin-sense transgenic mice [67]. Additionally, carbachol-induced phosphorylation of PKC-α was also elevated in these animals in comparison with WT mice suggesting that adiponectin increases calcium dependency of DSM contractions mediated by PKC-α expression.
It was previously shown that reduced expression of PKC in decompensated rabbit bladders was associated with increased frequency, decreased void, and consequently large residual volumes [25]. Additional cystometric studies in awake rats also revealed that pharmacological inhibition of PKC by Bim-1, and Ro318220 induced an increase in non-voiding contractions, frequency, and decreased void volume [22]. Interestingly, both inhibitors of PKC significantly reduced the ability to maintain muscle force of DSM but did not affect the sensitivity of force generation in response to EFS-mediated nerve stimulation. These data indicate that resting and low levels of PKC activity do not have a significant effect on nerve-mediated contractions in DSM. The ability to maintain DSM force is largely a function of the smooth muscle, and involves calcium sensitization mechanisms mediated by PKC, and Rho-kinase in a calcium-independent manner [3, 8, 24, 68]. Therefore, the frequency of micturition induced by reduced expression of PKC in decompensated rabbit bladders, and pharmacologic inhibition during cystometry in rat bladders are largely myogenic. The inability to maintain muscle force due to significantly low levels of PKC activity results in reduced bladder emptying, increased residual volumes, and an overall shortening of the micturition cycle leading to voiding frequency [22].
Partial bladder outlet obstruction, well known for its prevalence in aging males and often secondary to benign prostatic hyperplasia, has also been shown to alter structural proteins within the bladder wall leading to reduced compliance, DO, and frequency of urination. Among the structural proteins altered in PBOO are smooth muscle myosin (SMM) [84] collagen [85], caldesmon [80] and connective tissue [86]. A change in the ratio and organization of smooth muscle to non-smooth muscle elements is generally believed to lead to stiffening of the bladder wall, and a reduction in compliance resulting in a reduced storage capacity, and frequency of urination [77, 80, 85, 87]. However, significant changes in structural elements such as connective tissue, and collagen may represent some of more severe consequences of acute bladder outlet obstruction, associated with bladder decompensation, and may not represent some of the more subtle changes in bladder function that occur due to changes in regulatory proteins and associated with bladder compensation [3, 25, 88, 89].
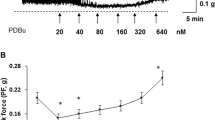
Figure 3 highlights the changes which characterize a milder form of dysfunction in compensated bladders such as decreased relaxation and increased DSM tone in response to the PKC activator, PDBu. Low levels of PKC stimulation which induce inhibition in normal DSM (panel A1) resulted in increased force in compensated DSM (panel A2). Additionally, moderate to high levels of PKC stimulation caused a modulated rise in tension in normal bladder DSM (panel B1), but a linear increase in the compensated DSM (panel B2). A failure to relax sufficiently and a linear, instead of a modular, increase in DSM tone are the contributing factors to a higher bladder pressure and bladder dysfunction observed in compensated bladders in comparison with normal and sham-operated controls.

Low and high levels of PKC stimulation by PDBu differentially affect DSM tone and spontaneous contractions. a Representative isolated muscle strips showing the effect of low (20nM) PDBu stimulation on sham (upper trace) and PBOO (lower trace) DSM strips from rabbits. Summary data is presented in c. b Representative isolated muscle strips showing the effect of high (100 nM) PDBu stimulation on sham (upper trace) and PBOO (lower trace). Summary data is presented in d. Low PDBu stimulation caused a significant inhibition of spontaneous contraction amplitude (SCA) of the sham muscle strips but only a minor effect in PBOO strips, however low PDBu caused a significant increase in basal DSM tone in the PBOO strips. High PDBu stimulation caused a significant increase in basal DSM tone for both sham and PBOO, however, there was a qualitative difference in the profile of the contractions. The force increase in the sham bladders displayed a biphasic effect whereas an increase in PBOO muscle strips tended to be more linear. High PDBu also significantly reduced the amplitude of spontaneous contractions for both sham and PBOO muscle strips
Study limitations
We acknowledge that our study has several limitations. First, we were not able to find any direct data on the role of PKC signaling in detrusor underactivity nor whether PKC pathways could be pharmacologically targeted to improve this condition. Second, we could not find sufficient information on the role of PKC pathways in sensory and efferent innervation of the lower urinary tract. This could be especially important for understanding the role of PKC in other pathological urologic conditions such as chronic pelvic pain (CPP) and bladder pain syndrome (BPS) [90, 91]. For example, a high level of PKC expression and/or activity has been suggested to play a role in feline interstitial cystitis [73], a naturally occurring bladder pain and dysfunctional voiding in cats. It would be of interest to determine whether or not PKC is overexpressed in these bladders, since high PKC stimulation was shown to increase bladder wall tension, and enhance neuronal sensitivity and contractility [8, 22, 25, 28]. Third, future studies focused on the role of PKC in the bladder urothelium are required to clarify the role of epithelial PKC in micturition.
Conclusions
PKC function in the urinary bladder extends far beyond its contribution, or the lack thereof, to maximum force generation in response to agonists, either directly, or in response to EFS. Data indicate that PKC is involved in the regulation of normal bladder function, and that PKC dysfunction is associated with DO, reduced contractility, and decreased void volume. Relaxation and modulation of spontaneous myogenic, and NVC via PKC-dependent activation of BK and KATP channels may also be one of its primary functions in support of bladder storage. Both in vitro, and in vivo studies reveal that PKC may be involved in regulating neuronal activity at the peripheral level in a concentration-dependent manner that may function in a complementary way to facilitate voiding. Finally, the demonstration that PKC dysfunction coexists with pathological changes such as PBOO, DO and is associated with reduced contractility and bladder emptying is further evidence of a significant and important role for PKC in the regulation of urinary bladder function.
Abbreviations
- Bim-1:
-
Bisindilylmaleimide1
- BK:
-
Large conductance calcium-activated potassium channel
- BSM:
-
Bladder smooth muscle
- BPS:
-
Bladder pain syndrome
- Ca++ :
-
Calcium
- CPP:
-
Chronic pelvic pain
- CNS:
-
Central nervous system
- DSM:
-
Detrusor smooth muscle
- DO:
-
Detrusor overactivity
- EFS:
-
Electrical field stimulation
- IF:
-
Integral force
- M3 :
-
Muscarinic
- MLCP:
-
Myosin light chain phosphorylation
- NVC:
-
Non-voiding contractions
- OAB:
-
Overactive bladder
- PBOO:
-
Partial bladder outlet obstruction
- PDBu:
-
Phorbol-12,13-dibutyrate
- PF:
-
Peak force
- PLC:
-
Phospholipase C
- PMA:
-
Phorbol-12,13-myristate
- PKC:
-
Protein kinase C
- ROK:
-
Rho-associated kinase
- VGCC:
-
Voltage gated calcium channels
- SK:
-
Small conductance ca++-activated potassium channels
- SMM:
-
Smooth muscle myosin
- THR:
-
Threonine
References
Andersson KE. Changes in bladder tone during filling: pharmacological aspects. Scand J Urol Nephrol Suppl. 1999;201:67–72. discussion 76–99.
Andersson KE. Detrusor myocyte activity and afferent signaling. Neurourol Urodyn. 2010;29(1):97–106.
Boopathi E, Hypolite JA, Zderic SA, Gomes CM, Malkowicz B, Liou HC, et al. GATA-6 and NF-kappaB activate CPI-17 gene transcription and regulate Ca2+ sensitization of smooth muscle contraction. Mol Cell Biol. 2013;33(5):1085–102.
Brading AF. Spontaneous activity of lower urinary tract smooth muscles: correlation between ion channels and tissue function. J Physiol. 2006;570(Pt 1):13–22.
Drake MJ, Harvey IJ, Gillespie JI. Autonomous activity in the isolated guinea pig bladder. Exp Physiol. 2003;88(1):19–30.
Gillespie JI. Modulation of autonomous contractile activity in the isolated whole bladder of the guinea pig. BJU Int. 2004;93(3):393–400.
Hypolite JA, Chang S, LaBelle E, Babu GJ, Periasamy M, Wein AJ, et al. Deletion of SM-B, the high ATPase isoform of myosin, upregulates the PKC-mediated signal transduction pathway in murine urinary bladder smooth muscle. Am J Physiol Renal Physiol. 2009;296(3):F658–665.
Hypolite JA, Lei Q, Chang S, Zderic SA, Butler S, Wein AJ, et al. Spontaneous and evoked contractions are regulated by PKC-mediated signaling in detrusor smooth muscle: involvement of BK channels. Am J Physiol Renal Physiol. 2013;304(5):F451–462.
Hypolite JA, Wein AJ, Haugaard N, Levin RM. Role of substrates in the maintenance of contractility of the rabbit urinary bladder. Pharmacology. 1991;42(4):202–10.
Levin RM, Hypolite J, Longhurst PA, Wein AJ. Comparison of the contractile and metabolic effects of muscarinic stimulation with those of KCl. Pharmacology. 1991;42(3):142–50.
Levin RM, Ruggieri MR, Velagapudi S, Gordon D, Altman B, Wein AJ. Relevance of spontaneous activity to urinary bladder function: an in vitro and in vivo study. J Urol. 1986;136(2):517–21.
Wein AJ. Physiology of micturition. Clin Geriatr Med. 1986;2(4):689–99.
Zhao Y, Wein AJ, Levin RM. Role of calcium in mediating the biphasic contraction of the rabbit urinary bladder. Gen Pharmacol. 1993;24(3):727–31.
Schneider T, Fetscher C, Krege S, Michel MC. Signal transduction underlying carbachol-induced contraction of human urinary bladder. J Pharmacol Exp Ther. 2004;309(3):1148–53.
Frazier EP, Peters SL, Braverman AS, Ruggieri Sr MR, Michel MC. Signal transduction underlying the control of urinary bladder smooth muscle tone by muscarinic receptors and beta-adrenoceptors. Naunyn Schmiedebergs Arch Pharmacol. 2008;377(4–6):449–62.
Frazier EP, Braverman AS, Peters SL, Michel MC, Ruggieri Sr MR. Does phospholipase C mediate muscarinic receptor-induced rat urinary bladder contraction? J Pharmacol Exp Ther. 2007;322(3):998–1002.
Durlu-Kandilci NT, Brading AF. Involvement of Rho kinase and protein kinase C in carbachol-induced calcium sensitization in beta-escin skinned rat and guinea-pig bladders. Br J Pharmacol. 2006;148(3):376–84.
Fleichman M, Schneider T, Fetscher C, Michel MC. Signal transduction underlying carbachol-induced contraction of rat urinary bladder. II. Protein kinases. J Pharmacol Exp Ther. 2004;308(1):54–8.
An JY, Yun HS, Lee YP, Yang SJ, Shim JO, Jeong JH, et al. The intracellular pathway of the acetylcholine-induced contraction in cat detrusor muscle cells. Br J Pharmacol. 2002;137(7):1001–10.
Aburto T, Jinsi A, Zhu Q, Deth RC. Involvement of protein kinase C activation in alpha 2-adrenoceptor-mediated contractions of rabbit saphenous vein. Eur J Pharmacol. 1995;277(1):35–44.
Dessy C, Kim I, Sougnez CL, Laporte R, Morgan KG. A role for MAP kinase in differentiated smooth muscle contraction evoked by alpha-adrenoceptor stimulation. Am J Physiol. 1998;275(4 Pt 1):C1081–1086.
Hypolite JA, Chang S, Wein AJ, Chacko S, Malykhina AP. Protein kinase C modulates frequency of micturition and non-voiding contractions in the urinary bladder via neuronal and myogenic mechanisms. BMC Urol. 2015;15(1):34.
Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50(2):279–90.
Wang T, Kendig DM, Smolock EM, Moreland RS. Carbachol-induced rabbit bladder smooth muscle contraction: roles of protein kinase C and Rho kinase. Am J Physiol Renal Physiol. 2009;297(6):F1534–1542.
Chang S, Hypolite JA, Mohanan S, Zderic SA, Wein AJ, Chacko S. Alteration of the PKC-mediated signaling pathway for smooth muscle contraction in obstruction-induced hypertrophy of the urinary bladder. Lab Invest. 2009;89(7):823–32.
Leiria LO, Sollon C, Calixto MC, Lintomen L, Monica FZ, Anhe GF, et al. Role of PKC and CaV1.2 in detrusor overactivity in a model of obesity associated with insulin resistance in mice. PLoS One. 2012;7(11):e48507.
Stanton MC, Austin JC, Delaney DP, Gosfield A, Marx JO, Zderic SA, et al. Partial bladder outlet obstruction selectively abolishes protein kinase C induced contraction of rabbit detrusor smooth muscle. J Urol. 2006;176(6 Pt 1):2716–21.
Wang T, Kendig DM, Trappanese DM, Smolock EM, Moreland RS. Phorbol 12,13-dibutyrate-induced, protein kinase C-mediated contraction of rabbit bladder smooth muscle. Front Pharmacol. 2012;2:83.
Ekman M, Andersson KE, Arner A. Receptor-induced phasic activity of newborn mouse bladders is inhibited by protein kinase C and involves T-type Ca2+ channels. BJU Int. 2009;104(5):690–7.
Somogyi GT, Tanowitz M, Zernova G, de Groat WC. M1 muscarinic receptor-induced facilitation of ACh and noradrenaline release in the rat bladder is mediated by protein kinase C. J Physiol. 1996;496(Pt 1):245–54.
Hristov KL, Smith AC, Parajuli SP, Malysz J, Petkov GV. Large-conductance voltage- and Ca2 + −activated K+ channel regulation by protein kinase C in guinea pig urinary bladder smooth muscle. Am J Physiol Cell Physiol. 2014;306(5):C460–470.
Petkov GV. Central role of the BK channel in urinary bladder smooth muscle physiology and pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2014;307(6):R571–584.
Huster M, Frei E, Hofmann F, Wegener JW. A complex of Ca(V)1.2/PKC is involved in muscarinic signaling in smooth muscle. FASEB J. 2010;24(8):2651–9.
Liu SH, Lin-Shiau SY. Protein kinase c regulates purinergic component of neurogenic contractions in mouse bladder. J Urol. 2000;164(5):1764–7.
Weng TI, Chen WJ, Liu SH. Bladder instillation of Escherichia coli lipopolysaccharide alters the muscle contractions in rat urinary bladder via a protein kinase C-related pathway. Toxicol Appl Pharmacol. 2005;208(2):163–9.
Petkov GV. Role of potassium ion channels in detrusor smooth muscle function and dysfunction. Nat Rev Urol. 2012;9(1):30–40.
Schneider T, Hein P, Michel MC. Signal transduction underlying carbachol-induced contraction of rat urinary bladder. I. Phospholipases and Ca2+ sources. J Pharmacol Exp Ther. 2004;308(1):47–53.
Wuest M, Hiller N, Braeter M, Hakenberg OW, Wirth MP, Ravens U. Contribution of Ca2+ influx to carbachol-induced detrusor contraction is different in human urinary bladder compared to pig and mouse. Eur J Pharmacol. 2007;565(1–3):180–9.
Zderic SA, Sillen U, Liu GH, Snyder 3rd MC, Duckett JW, Gong C, et al. Developmental aspects of excitation contraction coupling of rabbit bladder smooth muscle. J Urol. 1994;152(2 Pt 2):679–81.
Buckner SA, Milicic I, Daza AV, Coghlan MJ, Gopalakrishnan M. Spontaneous phasic activity of the pig urinary bladder smooth muscle: characteristics and sensitivity to potassium channel modulators. Br J Pharmacol. 2002;135(3):639–48.
Uchida W, Masuda N, Shirai Y, Shibasaki K, Satoh N, Takenada T. The role of extracellular Ca2+ in carbachol-induced tonic contraction of the pig detrusor smooth muscle. Naunyn Schmiedebergs Arch Pharmacol. 1994;350(4):398–402.
Masters JG, Neal DE, Gillespie JI. The contribution of intracellular Ca2+ release to contraction in human bladder smooth muscle. Br J Pharmacol. 1999;127(4):996–1002.
Kajioka S, Nakayama S, McMurray G, Abe K, Brading AF. Ca(2+) channel properties in smooth muscle cells of the urinary bladder from pig and human. Eur J Pharmacol. 2002;443(1–3):19–29.
Levin RM, Hypolite J, Ruggieri MR, Longhurst PA, Wein AJ. Effects of muscarinic stimulation on intracellular calcium in the rabbit bladder: comparison with metabolic response. Pharmacology. 1989;39(2):69–77.
Herrera GM, Heppner TJ, Nelson MT. Regulation of urinary bladder smooth muscle contractions by ryanodine receptors and BK and SK channels. Am J Physiol Regul Integr Comp Physiol. 2000;279(1):R60–68.
Bonev AD, Nelson MT. ATP-sensitive potassium channels in smooth muscle cells from guinea pig urinary bladder. Am J Physiol. 1993;264(5 Pt 1):C1190–1200.
Gopalakrishnan M, Whiteaker KL, Molinari EJ, Davis-Taber R, Scott VE, Shieh CC, et al. Characterization of the ATP-sensitive potassium channels (KATP) expressed in guinea pig bladder smooth muscle cells. J Pharmacol Exp Ther. 1999;289(1):551–8.
Buckner SA, Milicic I, Daza A, Davis-Taber R, Scott VE, Sullivan JP, et al. Pharmacological and molecular analysis of ATP-sensitive K(+) channels in the pig and human detrusor. Eur J Pharmacol. 2000;400(2–3):287–95.
Ha JH, Lee KY, Kim WJ. Actions of potassium channel openers in rat detrusor urinae. J Korean Med Sci. 1993;8(1):53–9.
Edwards G, Henshaw M, Miller M, Weston AH. Comparison of the effects of several potassium-channel openers on rat bladder and rat portal vein in vitro. Br J Pharmacol. 1991;102(3):679–86.
Malmgren A, Andersson KE, Sjogren C, Andersson PO. Effects of pinacidil and cromakalim (BRL 34915) on bladder function in rats with detrusor instability. J Urol. 1989;142(4):1134–8.
Bonev AD, Nelson MT. Muscarinic inhibition of ATP-sensitive K+ channels by protein kinase C in urinary bladder smooth muscle. Am J Physiol. 1993;265(6 Pt 1):C1723–1728.
Frayer SM, Barber LA, Vasko MR. Activation of protein kinase C enhances peptide release from rat spinal cord slices. Neurosci Lett. 1999;265(1):17–20.
Braverman AS, Doumanian LR, Ruggieri Sr MR. M2 and M3 muscarinic receptor activation of urinary bladder contractile signal transduction. II. Denervated rat bladder. J Pharmacol Exp Ther. 2006;316(2):875–80.
Longhurst PA, Leggett RE, Briscoe JA. Characterization of the functional muscarinic receptors in the rat urinary bladder. Br J Pharmacol. 1995;116(4):2279–85.
Boberg L, Poljakovic M, Rahman A, Eccles R, Arner A. Role of Rho-kinase and protein kinase C during contraction of hypertrophic detrusor in mice with partial urinary bladder outlet obstruction. BJU Int. 2012;109(1):132–40.
Eto M, Ohmori T, Suzuki M, Furuya K, Morita F. A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterization. J Biochem. 1995;118(6):1104–7.
Eto M, Senba S, Morita F, Yazawa M. Molecular cloning of a novel phosphorylation-dependent inhibitory protein of protein phosphatase-1 (CPI17) in smooth muscle: its specific localization in smooth muscle. FEBS Lett. 1997;410(2–3):356–60.
Adelstein RS, Eisenberg E. Regulation and kinetics of the actin-myosin-ATP interaction. Annu Rev Biochem. 1980;49:921–56.
Ng YK, de Groat WC, Wu HY. Muscarinic regulation of neonatal rat bladder spontaneous contractions. Am J Physiol Regul Integr Comp Physiol. 2006;291(4):R1049–1059.
Hristov KL, Parajuli SP, Soder RP, Cheng Q, Rovner ES, Petkov GV. Suppression of human detrusor smooth muscle excitability and contractility via pharmacological activation of large conductance Ca2 + −activated K+ channels. Am J Physiol Cell Physiol. 2012;302(11):C1632–1641.
Brading AF. Ion channels and control of contractile activity in urinary bladder smooth muscle. Jpn J Pharmacol. 1992;58 Suppl 2:120P–7P.
Rivera L, Brading AF. The role of Ca2+ influx and intracellular Ca2+ release in the muscarinic-mediated contraction of mammalian urinary bladder smooth muscle. BJU Int. 2006;98(4):868–75.
Klockner U, Isenberg G. Action potentials and net membrane currents of isolated smooth muscle cells (urinary bladder of the guinea-pig). Pflugers Arch. 1985;405(4):329–39.
Nakamura Y, Ishiura Y, Yokoyama O, Namiki M, De Groat WC. Role of protein kinase C in central muscarinic inhibitory mechanisms regulating voiding in rats. Neuroscience. 2003;116(2):477–84.
Hristov KL, Chen M, Kellett WF, Rovner ES, Petkov GV. Large-conductance voltage- and Ca2 + −activated K+ channels regulate human detrusor smooth muscle function. Am J Physiol Cell Physiol. 2011;301(4):C903–912.
Nobe K, Fujii A, Saito K, Negoro T, Ogawa Y, Nakano Y, et al. Adiponectin enhances calcium dependency of mouse bladder contraction mediated by protein kinase Calpha expression. J Pharmacol Exp Ther. 2013;345(1):62–8.
Takahashi R, Nishimura J, Hirano K, Seki N, Naito S, Kanaide H. Ca2+ sensitization in contraction of human bladder smooth muscle. J Urol. 2004;172(2):748–52.
Yoshimura Y, Yamaguchi O. Calcium independent contraction of bladder smooth muscle. Int J Urol. 1997;4(1):62–7.
Boopathi E, Gomes C, Zderic SA, Malkowicz B, Chakrabarti R, Patel DP, et al. Mechanical stretch upregulates proteins involved in Ca2+ sensitization in urinary bladder smooth muscle hypertrophy. Am J Physiol Cell Physiol. 2014;307(6):C542–553.
Ji G, Barsotti RJ, Feldman ME, Kotlikoff MI. Stretch-induced calcium release in smooth muscle. J Gen Physiol. 2002;119(6):533–44.
Downing JE, Role LW. Activators of protein kinase C enhance acetylcholine receptor desensitization in sympathetic ganglion neurons. Proc Natl Acad Sci U S A. 1987;84(21):7739–43.
Sculptoreanu A, de Groat WC, Buffington CA, Birder LA. Protein kinase C contributes to abnormal capsaicin responses in DRG neurons from cats with feline interstitial cystitis. Neurosci Lett. 2005;381(1–2):42–6.
Zhou Y, Zhou ZS, Zhao ZQ. PKC regulates capsaicin-induced currents of dorsal root ganglion neurons in rats. Neuropharmacology. 2001;41(5):601–8.
Parajuli SP, Hristov KL, Cheng Q, Malysz J, Rovner ES, Petkov GV. Functional link between muscarinic receptors and large-conductance Ca-activated K channels in freshly isolated human detrusor smooth muscle cells. Pflugers Archiv. 2015;467(4):665-75. doi:10.1007/s00424-014-1537-8. Epub 2014 May 28.
Parajuli SP, Hristov KL, Cheng Q, Malysz J, Rovner ES, Petkov GV. Functional link between muscarinic receptors and large-conductance Ca2 + − activated K+ channels in freshly isolated human detrusor smooth muscle cells. Pflugers Arch. 2015;467(4):665–75.
Levin RM, Haugaard N, Hypolite JA, Wein AJ, Buttyan R. Metabolic factors influencing lower urinary tract function. Exp Physiol. 1999;84(1):171–94.
Levin RM, Monson FC, Haugaard N, Buttyan R, Hudson A, Roelofs M, et al. Genetic and cellular characteristics of bladder outlet obstruction. Urol Clin North Am. 1995;22(2):263–83.
Shahab N, Kajioka S, Takahashi-Yanaga F, Onimaru M, Matsuda M, Seki N, et al. Obstruction enhances rho-kinase pathway and diminishes protein kinase C pathway in carbachol-induced calcium sensitization in contraction of alpha-toxin permeabilized guinea pig detrusor smooth muscle. Neurourol Urodyn. 2012;31(4):593–9.
Zhang EY, Stein R, Chang S, Zheng Y, Zderic SA, Wein AJ, et al. Smooth muscle hypertrophy following partial bladder outlet obstruction is associated with overexpression of non-muscle caldesmon. Am J Pathol. 2004;164(2):601–12.
Choi BH, Jin LH, Kim KH, Kang SA, Kang JH, Yoon SM, et al. Cystometric parameters and the activity of signaling proteins in association with the compensation or decompensation of bladder function in an animal experimental model of partial bladder outlet obstruction. Int J Mol Med. 2013;32(6):1435–41.
Levin RM, Longhurst PA, Monson FC, Kato K, Wein AJ. Effect of bladder outlet obstruction on the morphology, physiology, and pharmacology of the bladder. Prostate Suppl. 1990;3:9–26.
Stein R, Gong C, Hutcheson JC, Canning DA, Zderic SA. The decompensated detrusor III: impact of bladder outlet obstruction on sarcoplasmic endoplasmic reticulum protein and gene expression. J Urol. 2000;164(3 Pt 2):1026–30.
DiSanto ME, Stein R, Chang S, Hypolite JA, Zheng Y, Zderic S, et al. Alteration in expression of myosin isoforms in detrusor smooth muscle following bladder outlet obstruction. Am J Physiol Cell Physiol. 2003;285(6):C1397–1410.
Macarak EJ, Schulz J, Zderic SA, Sado Y, Ninomiya Y, Polyak E, et al. Smooth muscle trans-membrane sarcoglycan complex in partial bladder outlet obstruction. Histochem Cell Biol. 2006;126(1):71–82.
Sjuve R, Haase H, Ekblad E, Malmqvist U, Morano I, Arner A. Increased expression of non-muscle myosin heavy chain-B in connective tissue cells of hypertrophic rat urinary bladder. Cell Tissue Res. 2001;304(2):271–8.
Iguchi N, Hou A, Koul HK, Wilcox DT. Partial bladder outlet obstruction in mice may cause E-cadherin repression through hypoxia induced pathway. J Urol. 2014;192(3):964–72.
Chacko S, Chang S, Hypolite J, Disanto M, Wein A. Alteration of contractile and regulatory proteins following partial bladder outlet obstruction. Scand J Urol Nephrol Suppl. 2004;215:26–36.
Chang S, Gomes CM, Hypolite JA, Marx J, Alanzi J, Zderic SA, et al. Detrusor overactivity is associated with downregulation of large-conductance calcium- and voltage-activated potassium channel protein. Am J Physiol Renal Physiol. 2010;298(6):F1416–1423.
Malykhina A, Hanno P. How are we going to make progress treating bladder pain syndrome? ICI-RS 2013. Neurourol Urodyn. 2014;33(5):625–9.
Sadler KE, Stratton JM, Kolber BJ. Urinary bladder distention evoked visceromotor responses as a model for bladder pain in mice. J Vis Exp. 2014:(86).
Acknowledgments
The study has been supported by the NIH/NIDDK grant DK095817.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JAH - conception and design, analysis and interpretation of the data, drafting of the manuscript. APM - general supervision, critical revision for intellectual content. All authors have made a significant contribution to the paper, and have read and approved the final draft.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hypolite, J.A., Malykhina, A.P. Regulation of urinary bladder function by protein kinase C in physiology and pathophysiology. BMC Urol 15, 110 (2015). https://doi.org/10.1186/s12894-015-0106-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12894-015-0106-6