
Abstract
Background
Kawasaki disease (KD) is a febrile systemic vasculitis involvingchildren younger than five years old. However, the specific biomarkers and precise mechanisms of this disease are not fully understood, which can delay the best treatment time, hence, this study aimed to detect the potential biomarkers and pathophysiological process of KD through bioinformatic analysis.
Methods
The Gene Expression Omnibus database (GEO) was the source of the RNA sequencing data from KD patients. Differential expressed genes (DEGs) were screened between KD patients and healthy controls (HCs) with the “limma” R package. Weighted gene correlation network analysis (WGCNA) was performed to discover the most corresponding module and hub genes of KD. The node genes were obtained by the combination of the least absolute shrinkage and selection operator (LASSO) regression model with the top 5 genes from five algorithms in CytoHubba, which were further validated with the receiver operating characteristic curve (ROC curve). CIBERSORTx was employed to discover the constitution of immune cells in KDs and HCs. Functional enrichment analysis was performed to understand the biological implications of the modular genes. Finally, competing endogenous RNAs (ceRNA) networks of node genes were predicted using online databases.
Results
A total of 267 DEGs were analyzed between 153 KD patients and 92 HCs in the training set, spanning two modules according to WGCNA. The turquoise module was identified as the hub module, which was mainly enriched in cell activation involved in immune response, myeloid leukocyte activation, myeloid leukocyte mediated immunity, secretion and leukocyte mediated immunity biological processes; included type II diabetes mellitus, nicotinate and nicotinamide metabolism, O-glycan biosynthesis, glycerolipid and glutathione metabolism pathways. The node genes included ADM, ALPL, HK3, MMP9 and S100A12, and there was good performance in the validation studies. Immune cell infiltration analysis revealed that gamma delta T cells, monocytes, M0 macrophage, activated dendritic cells, activated mast cells and neutrophils were elevated in KD patients. Regarding the ceRNA networks, three intact networks were constructed: NEAT1/NORAD/XIST-hsa-miR-524-5p-ADM, NEAT1/NORAD/XIST-hsa-miR-204-5p-ALPL, NEAT1/NORAD/XIST-hsa-miR-524-5p/hsa-miR-204-5p-MMP9.
Conclusion
To conclude, the five-gene signature and three ceRNA networks constructed in our study are of great value in the early diagnosis of KD and might help to elucidate our understanding of KD at the RNA regulatory level.
Similar content being viewed by others


Introduction
Kawasaki disease (KD), termed mucocutaneous lymph node syndrome, is a kind of febrile systematic vasculitis of unknown etiology, that affects children under 5 years old. The pathophysiological mechanisms of KD involve the ectopic activation of the immune system and overwhelming release of proinflammatory cytokines which can result in abnormalities in micro and medium vessels, especially coronary arteries (CA) [1, 2]. The standard regimen of intravenous immunoglobulin (IVIG) at 2 g/kg with aspirin intervention between Days 5–7 of the illness can significantly decrease the incidence of CA lesion (CAL), which is the most frequent ensuing catastrophic issue [3, 4], and could progress into CA aneurysm (CAA), stenosis, thrombosis, and myocardial infarction [2]. Unfortunately, delayed treatment and resistance to IVIG of 10%-20% of patients give rise to the development of CAA. The Current diagnosis of KD is mainly based on a collection of clinical features, including fever (usually ≥ 39 °C) for more than 5 days, strawberry tongue, cracked lips, bilateral conjunctivitis, cervical lymphadenopathy, edema of the extremities, general rash, and CAL detected on echocardiography. The adjunctive diagnoses mostly engage inflammation biomarkers, such as erythrocyte sedimentation rate, white blood cell/leukocyte count, platelet count, C-reactive protein, interleukin-6 (IL-6), and serum albumin levels [5, 6].
Bioinformatic analysis of microarray data is one of the first well-established methodologies for the high-throughput analysis of biological systems. With the advances in computer technology, many gene expression profiling studies of KD have been performed to discover its pathogenesis. Weighted Gene Coexpression Network Analysis (WGCNA) is an unsupervised clustering approach that focuses on the gross expression profile of genes to avoid gene loss, thus providing a comprehensive description of cellular responses and aiming to discover the associations between genes, modules and phenotypes [7, 8]. The LASSO regression model is a multigene-based classifier, which is a prevailing high-dimensional variable regression analysis algorithm [9]. The combination of WGCNA and LASSO represents a credible approach to reveal novel biomarkers. Additionally, the construction of competitive endogenous RNA (ceRNA) networks will help to illustrate the novel mechanisms of transcriptional regulation.
To date, articles about KD have mainly focused on the following aspects. First, it is difficult to differentiate KD from other febrile diseases, such as bacterial and viral infections and systemic lupus erythematosus (SLE), because of the atypical clinical symptoms and laboratory indicators. Previous papers have ascertained optimal signatures at different levels, including whole blood, specific cell type, urine peptidome and clinical information, to sequester KD from clinically confusing diseases to prompt timely treatment and to prevent the occurrence of complications [10,11,12,13]. Second, KD patients who are unresponsive to the administration of IVIG can develop CAA; therefore, the difference between IVIG-responsive and IVIG-resistant patients and many scoring systems predicting the responsiveness to IVIG have been explored [12, 14, 15]. In addition, a few bioinformatic analyses have been introduced to uncover potential biomarkers of KD [16,17,18,19]. For our study, we aimed to discover potential biomarkers for the early diagnosis and treatment of KD. This study is the first to integrated WGCNA, LASSO regression and CytoHubba in Cytoscape to predict node genes of KD with the combination of two datasets from the public database. One independent dataset of KD pre- and post- IVIG therapy was gathered for further validation. In addition, the relationships between the infiltration of immune cells and node genes were analyzed to scale up our understanding of KD from the perspective of molecular immune mechanisms. Furthermore, the establishment of ceRNA networks of node genes would elucidate the mechanism of KD at the RNA regulatory level. In summary, this study that hybridized the aforementioned bioinformatic analysis might have the capability of providing candidate biomarkers for the early diagnosis and treatment of KD.
Material and methods
Microarray data
In Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) database, four publicly available datasets related to Kawasaki disease (KD) were retrieved, and no further approval from the local ethics committee was required. GSE68004 [20] and GSE73461 [10] were both based on GPL10558 Illumina HumanHT-12 V4.0 expression beadchip platform. GSE68004 includes 76 KD patients and 37 health controls (HC), and GS73461 contains 77 KD patients and 55 HC (the information of one KD patient was missed). Because of the similar sample size and the same platform, these two datasets were incorporated to perform combined analysis, which was defined as the training set. GSE18606 [21] was assayed on GPL6480 Agilent-014850 Whole Human Genome Microarray, which was regarded as the validation set, and 20 KD patients and 9 HC were chosen. GSE63881 [12] was set as the validation set for KD patients following IVIG, based on the same platform as the training set and covering 171 acute (before IVIG) and 170 convalescent (after IVIG) KD patients. The detailed information of the four datasets was shown in Table 1.
Identification of differentially expressed genes (DEGs) by combined analysis
GSE68004 and GSE73461 were combined with the “inSilicoMerging” package [22]. Then batch normalization was performed through the ComBat method in the “sva” R package [23]. A platform annotation file was used to convert the probe expression into a gene expression matrix, and noncoding RNAs (ncRNAs) were removed from this matrix. DEGs between KD and HC were acquired by the eBayes function in the “limma” package [24]. A heatmap of the top ten upregulated and top ten downregulated genes was generated between KDs and HCs using the “pheatmap” package in R [25].
Weighted gene coexpression network analysis (WGCNA)
We applied WGCNA to explore the interactions between genes and genes and between genes and clinical traits. The expression profile of the DEGs from the combined analysis was utilized to conduct WGCNA through the “WGCNA package” in R [7]. First, the hclust function was used to assess the presence of any obvious outliers and cluster the samples by a hierarchical clustering algorithm. Second, the optimal soft threshold for the adjacency matrix was selected with the pickSoftThreshold and softConnectivity functions so that the constructed correlation network was more approximate to the scale-free characteristic for the biological network. Third, hierarchical clustering and dynamic tree cut functions were implemented to identify gene modules that could sectionalize genes with the same expression pattern, and a minimum module size of 30 was adopted. The fourth step was to connect modules with the clinical traits, and in this study, we explored the relationships of the modules with disease and sex. Finally, gene significances (GS) and module memberships (MM) were obtained to recognize the hub genes with cutoffs of GS > 0.7 and MM > 0.9. GS was calculated to quantify the absolute value of associations between individual genes and the trait of interest (disease and sex in this analysis), while MM was the correlation of the module eigengene and gene expression profile. Module significance (MS), the average GS across genes in the module, was used to identify the hub module. Therefore, it is feasible to obtain hub genes in the hub module with GS and MM. Genes in the hub module were then retrieved to perform functional enrichment analysis with “clusterProfiler” R package [26] and the categories including biological process (BPs) in Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were selected [27]. p-value < 0.05 was considered to be criteria of significant enrichment. The top 10 BPs and KEGG pathways are displayed in the form of bubble plots with the aid of the “ggplot2” R package [28].
Construction of a protein–protein interaction (PPI) network
The STRING database (http://string-db.org) [29] was used to identify known and predicted PPIs. Genes in the hub module were imported into STRING, and the interaction file was acquired and input into Cytoscape (version 3.7.2) for better visualization. The CytoHubba plugin of Cytoscape [30] was applied to score the top 5 genes by four algorithms incorporating MCC (maximal clique centrality), MNC (maximum neighborhood component), EPC (edge percolated component) degree and Betweenness. Genes that appeared in the top 5 genes of the five algorithms were retained.

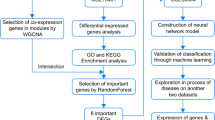
The workflow of the present study
Construction of least absolute shrinkage and selection operator (LASSO) model
LASSO is a penalized regression method that uses the L1 penalty to restrain regression coefficients (λ) toward zero, and genes with nonzero coefficients are retained as seed genes. The expression profile of hub genes was transferred to the R platform to perform LASSO regression analysis, which is based on the “glmnet” package [9] with nfolds = 10. The best λ-value was selected according to the minimum criterion. The model index for each sample was constituted with the following equation:
The “Coef” indicates the regression coefficient of the gene that is rooted in the LASSO regression to weight the expression value of the selected gene, and “Exp” implies the expression value of the gene.
Validation of the selected genes in datasets
ROC curves of all the selected genes in the training set and GSE18606, including genes retained in the LASSO model and the algorithms in the CytoHubba, were plotted with the roc function in the “pROC” package [31], and genes with area under the curve (AUC) > 0.9 in both sets were defined as node genes. Then batch effect of node genes between different platforms of the training set and validation set was eliminated using the “sva” R package. The difference in node genes was additionally investigated in GSE63881 to scrutinize the expression change following IVIG. GraphPad Prism (version 8.0.2) was used to construct violin plots of node genes in the three datasets, p–values between groups were calculated with the Wilcoxon test in GSE18606 and GSE63881. Principal component analysis (PCA) plots were used to display the efficacy of the node-gene signature to distinguish KDs from HCs, which were analyzed and visualized using the “limma” and “ggplot2” packages, respectively.

Weighted gene correlation network analysis (WGCNA) and functional annotations of the hub genes. A The heatmap of top 10 up- and down-regulated genes between Kawasaki disease (KD) patients and health controls (HC). Row: genes, Columns: samples. Colors indicate the gene expression level, in which red means high level and blue means low level. B Cluster dendrogram represents the distribution of genes with corresponding module colors, which incorporates a sum of 2 modules, and genes that don’t co-express with other genes are divided into the gray module. C Module-trait correlation heatmap. Numbers in the upper left corners represent the correlation coefficient of modules to traits, red color represents positive correlation, and green color represents negative correlation. Numbers in the lower right corners means the p-values. Each row symbolizes a module eigengene and each column symbolizes a trait. Sex(F): female; Sex(M): male. D The bubble plot displays the significant enriched biological processes (BPs) of 80 genes. E The bubble plot shows the significant enriched pathways of 80 genes. The color of the dots refers to the -log10 (p-value), and the size of the dots refers to the number of DEGs mapped to the indicted pathways, respectively. The significant biological processes and pathways are selected according to p-value < 0.05

Screening of node genes. A PPI network map of 80 genes, which includes 33 nodes and 43 edges. Light brown circles stand for common genes appearing in the five algorithms. B LASSO model. λ = λ.1se, mean-squared error (MSE) = 0.02655. C The variation of coefficients of hub genes with different Log λ. ROC curves of the seven genes alone (ADM, ALPL, BCL6, HK3, MMP9, PYGL and S100A12) in both the training set (D) and validation set (GS18606) (E)

Validation of node genes. A Violin plots of node genes between KD and HC in training set. B Violin plots of node genes between KD and HC in GSE18606. C Violin plots of node genes between A and C in GSE63881. Pink represents KD, blue represents HC, deep pink refers to A, and light pink refers to C. (****P < 0.0001). “A” and “C” means acute and convalescent KD patients, separately

Immune cell infiltration and correlation with genes. A Comparison of 22 immune cell subtypes between KDs and HCs. p-values are obtained by the Wilcoxon test. Pink and turquoise color represent KDs and HCs, respectively (_: non-significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001) (The fraction of eosinophils is zero.). The immune cells displayed with red color are upregulated prominently in KD. Correlation between ADM and neutrophils (B); ALPL and neutrophils (C); HK3 and neutrophils (D); MMP9 and neutrophils (E); S100A12 and neutrophils (F). “r” means the correlation coefficient between nodes and immune cells

Prediction of ceRNA network. A The miRNA-mRNA interaction network includes 79 nodes and 77 edges. The ceRNA networks of NEAT1/NORAD/XIST-hsa-miR-524-5p-ADM (B), NEAT1/NORAD/XIST-hsa-miR-204-5p-ALPL (C), and NEAT1/NORAD/XIST-hsa-miR-524-5p/hsa-miR-204-5p-MMP9 (D). Red circles stand for node genes. Triangles stand for miRNAs (brown triangles: connect with only one mRNA; yellow triangles: connect with two mRNAs). And green Vs represent lncRNAs
Immune cell infiltration analysis
To prove the difference in the relative proportions of infiltrating immune cells between KDs and HCs, a bioinformatic algorithm called CIBERSORTx (https://cibersortx.stanford.edu/) [32] was used. The abundance level of immune cells was assessed using the 22 kinds of immune cell subsets with 1000 permutations. GraphPad Prism (version 8.0.2) [33] and the Wilcoxon test were utilized to display and analyze the comparative expression of immune cells. A p-value < 0.05 was regarded as the criterion for a significant difference. The correlation analysis among immune cells and node genes was undertaken using the “corrplot” R package [34]. The relationships between node genes and the most relevant immune cells were determined by Spearman correlation analysis and are shown using the “ggplot2” package.
Prediction of ncRNAs
MiRNAs targeting node genes were predicted by the miRTarBase database in the Enrichr platform (http://amp.pharm.mssm.edu/Enrichr/) [35] with the threshold of p–value < 0.05. The lncRNAs interacting with the remaining miRNAs were predicted through starBase (http://starbase.sysu.edu.cn/) [36], with the following criteria: mammalian, human h19 genome, CLIP-Data ≥ 5, and with or without degradome data. The coexpression networks of mRNAs-miRNAs and ceRNA networks based on mRNAs-miRNAs-lncRNAs were established and visualized utilizing Cytoscape (version 3.7.2).
Statistical analysis
DEGs between KDs and HCs were defined by p-values < 0.05 and |log2 fold change (FC)|≥ 2. The Wilcoxon test was applied to compare the mRNA expression in the datasets and the proportions of immune cells between KDs and HCs, and p-values < 0.05 were considered to be significant. The cutoff value for the selection of BPs and KEGG pathways was p-value < 0.05. ROC curve analysis and PCA were performed to assess the diagnostic accuracy and value of the selected genes. The relationship between node genes and immune cells was calculated through Spearman’s rank correlation. And p-values < 0.05 were considered statistically significant for almost all results, and all tests were two-tailed. The visualization of the data was accomplished by R 4.0.5, GraphPad Prism (version 8.0.2) and Cytoscape (version 3.7.2).
Results
Identification of DEGs by combined analysis
Based on the workflow in Fig. 1, we concluded that after combined analysis of datasets GSE68004 and GSE73461, there were 267 DEGs with p-values < 0.05 and |log2 FC|≥ 2, in which 231 were upregulated and 36 were downregulated in KD. The top 10 upregulated and downregulated DEGs are presented in the heatmap (Fig. 2A).
Modules associated with KD
The soft threshold power (β) of 28 was the optimal power, while the scale-free topology fit index was 0.86 along with a mild mean-connectivity (Figure S1), which made the network accord with the power-law distribution in WGCNA. Then, a total of two modules were finally obtained (Fig. 2B-C), of which the turquoise module exhibited the highest biological association with KD (correlation coefficient = 0.73, P = 9.5E-42; Fig. 2C), and we declared the turquoise module to be the hub module. No significant linkage was found between the module and the sex of the samples. The hub module contained a total of 80 DEGs, and nine genes upregulated in KD were selected as centrally located intramodular hub genes (ACSL1, ADM, ALPL, B4GALT5, BCL6, DYSF, LOC440731, PYGL, TLR5) with the cutoff criteria of GS > 0.7 and MM > 0.9. In addition, genes that could not be co-expressed with other genes were assigned to the grey module, which was ignored in our study. According to the enrichment analysis of the hub module, the biological processes (Fig. 2D) were mainly enriched in cell activation involved in immune response, myeloid leukocyte activation, myeloid leukocyte mediated immunity, secretion and leukocyte mediated immunity; the prominent pathways (Fig. 3E) included type II diabetes mellitus, nicotinate and nicotinamide metabolism, O-glycan biosynthesis, glycerolipid and glutathione metabolism.
Filtration and validation of node genes
For the sake of screening node genes that contribute to KD, we not only constructed the PPI network of genes in the hub module, but also identified the seed genes among the hub genes through LASSO regression analysis. The PPI network of genes acquired from WGCNA was established, with 33 nodes and 43 edges (Fig. 3A). Then the top 5 genes identified by the five algorithms in CytoHubba were crossed to obtain the common genes (Table 2), with the results of S100A12, HK3 and MMP9. Besides, the LASSO model was constructed (Fig. 3B-C) with four genes (ADM, ALPL, BCL6 and PYGL) possessing non-zero coefficients when λ.1se = 0.17130. Detailed information of the results is displayed in Table 3. To confirm the efficiency of both the algorithms in CytoHubba and the LASSO model, ROC curve analysis was performed (training set: the AUC of ADM = 0.977, the AUC of ALPL = 0.979, the AUC of BCL6 = 0.975, and the AUC of HK3 = 0.980, the AUC of MMP9 = 0.965, the AUC of PYGL = 0.977, the AUC of S100A12 = 0.971, Fig. 3D; GSE18606: the AUC of ADM = 0.950, the AUC of ALPL = 0.994, the AUC of BCL6 = 0.872, and the AUC of HK3 = 0.972, the AUC of MMP9 = 0.956, the AUC of PYGL = 0.900, the AUC of S100A12 = 0.978, Fig. 3E). The node genes were acquired according to their AUC values in two datasets, which incorporated ADM, ALPL, HK3, MMP9 and S100A12. For validation, violin plots which presented clearly separated trend (Fig. 4A-B) in the two datasets, and PCA (Figure S2A-B) suggesting a striking difference between KDs and HCs were conducted. In short, all of these genes effectively distinguish KDs from HCs, which also indicated the prominent predictive precision of both the LASSO model and algorithms in CytoHubba. In addition, these genes were markedly downregulated following IVIG treatment (Fig. 4C), which demonstrated that node genes were related to immune and inflammatory responses in KD and might be potential therapeutic targets for IVIG treatment.
Immune cell infiltration and correlation analysis with node genes
To obtain a deeper understanding of the mechanisms of KD, we explored and compared the composition of the infiltrating immune cells between KDs and HCs in the training set (Fig. 5A), eosinophils had an estimated abundance of 0). The proportions of gamma delta T cells (P < 0.000001), monocytes (P < 0.000001), M0 macrophages (P < 0.000001), activated dendritic cells (P = 0.00082), activated mast cells (P < 0.000001) and neutrophils (P < 0.000001) were higher in KDs. However, the fractions of memory B cells (P < 0.000001), plasma cells (P = 0.00082), CD8+ T cells (P < 0.000001), CD4+ naïve T cells (P = 0.000011), CD4+ resting memory T cells (P < 0.000001), CD4+ activated memory T cells (P = 0.00027), resting NK cells (P < 0.000001), activated NK cells (P < 0.000001), M2 macrophages (P < 0.000001), resting dendritic cells (P = 0.03) and resting mast cells (P < 0.000001) were higher in HCs. We then computed the correlation among the five node genes (ADM, ALPL, HK3, MMP9 and S100A12) and immune cells, as shown in Figure S3. The correlation between node genes and neutrophils, that were most positively associated with node genes, were illustrated as follows: ADM (r = 0.62, P < 0.0001, Fig. 5B), ALPL (r = 0.66, P < 0.0001, Fig. 5C), HK3 (r = 0.64, P < 0.0001, Fig. 5D), MMP9 (r = 0.64, P < 0.0001, Fig. 5E) and S100A12 (r = 0.57, P < 0.0001, Fig. 5F).
Prediction of ncRNAs and construction of ceRNA networks
MiRNAs can induce gene degradation by binding to the 3’UTR of target mRNAs, and lncRNAs can competitively bind miRNAs, which are known as competing endogenous RNAs (ceRNAs). Both miRNAs and lncRNAs have been claimed to be involved in KD [44, 46, 47]; hence, we analyzed the target lncRNAs of the miRNAs interacting with node genes through online databases. The mRNA‒miRNA pairs were presented in Fig. 6A and Table 4, with 79 nodes and 77 edges. The top 2 most significant miRNAs were hsa-miR-524-5p (p-value = 0.000723) and hsa-miR-204-5p (p-value = 0.003814884), and they connected with two node genes. On the basis of starBase, the co-associative lncRNAs are NEAT1, NORAD and XIST. A total of three ceRNA networks were ultimately constructed: NEAT1/NORAD/XIST-hsa-miR-524-5p-ADM (Fig. 6B), NEAT1/NORAD/XIST-hsa-miR-204-5p-ALPL (Fig. 6C), NEAT1/NORAD/XIST-hsa-miR-524-5p/hsa-miR-204-5p-MMP9 (Fig. 6D).
Discussion
KD is a self-limited multisystemic vasculitis that develops in genetically susceptible children after exposure to stimuli, mostly microbial and viral infections. Marked activation of immune and inflammatory reactions plays important roles in the development of KD [37, 38]. IVIG, which is a plasma-derived polyclonal IgG preparation, is considered the first-line therapy for KD and can strikingly decrease the incidence of CAA. However, without specific biomarkers, it is difficult to diagnose and give timely treatment at the early stage, which elevates the probability of CAA. Accordingly, the purpose of this research was to identify potential diagnostic biomarkers and possible mechanisms of KD through bioinformatic analysis and to offer an original direction for recognizing and treating KD patients.
We mainly combined two bioinformatic analyses to identify the underlying biomarkers of KD. First, WGCNA is a system biology approach which can group genes into modules with the same expression patterns, and correlate clinical features with modules. Compared with concentrating on DEGs, WGCNA can use the information of thousands of genes with the greatest variations to identify the interesting gene sets [39]. Many kinds of cancer such as breast [40] and gastric cancer [41], and other diseases like KD [18, 42] have been analyzed by WGCNA. And in this study, the turquoise module was identified as the hub module according to the results of WGCNA, and the included genes were subjected to clusterprofiler R package. The dominant enriched BP terms in the GO analysis were cell activation involved in immune response, myeloid leukocyte activation, myeloid leukocyte mediated immunity, secretion and leukocyte mediated immunity biological processes [48, 49]; the prominent pathways included type II diabetes mellitus, nicotinate and nicotinamide metabolism, O-glycan biosynthesis, glycerolipid and glutathione metabolism.
Second, LASSO regression analysis is a complex algorithm, that uses regularization to enhance the accuracy of prediction. And it was applied to the expression matrix of candidate genes to construct the prognostic multi-gene signature. Multiple RNA risk signatures were identified through LASSO penalized Cox regression analysis [43, 44]. Besides, the combination of these two methods has become a tendency recently, characteristic genes were determined in endometriosis [45] and sepsis [46, 47] with the application of the combination of WGCNA and LASSO. Regarding the gene signature of our analysis, a five-gene signature was formed after combining the top five genes of the five algorithms in CytoHubba and the retained genes in LASSO analysis, comprising ADM, ALPL, HK3, MMP9 and S100A12, all were significantly upregulated in KD and with good validation performance in other cohorts.
Adrenomedullin (ADM) is a vasoactive peptide mediating vasodilation and endothelial function regulation. The expression level of ADM has been reported to be elevated in KD [48, 49], and higher in KD patients developed CAAs [48]. Plasma ADM levels are positively correlated to the degree of endothelial damage in atherosclerosis patients [50], which indicate that it is a good indicator of the prognosis in patients with CA disease [51]. Alkaline phosphatase (ALPL), which could regulate tissue mineralization and execute an integral function in cardiovascular remodeling [52], has been identified to be increased in acute KD patients and decreased after IVIG infusion [53], which are concordant with our analysis. In addition, ALPL could predict the morbidity and mortality of cardiovascular-related disease [54, 55], and is associated with inflammation [56]. Hexokinase 3 (HK3) is involved in the glucose metabolic pathway and also correlated with immune cell infiltration [57]. HK3 has been detected to be upregulated in IVIG-non responders of KD [58], and further examination should be conducted in both IVIG-non responders and responders of acute KD. Matrix metalloproteinase-9 (MMP9), a member of MMPs family, could degrade extracellular matrix components and assume an important role in the processes of inflammation and tissue remodeling. Elevated expression of MMP9 has been reported in acute KD [59] through mediating vascular smooth muscle cell (VSMC) migration and neointimal formation, besides, it is also related to the formation of CAA in KD [60,61,62]. S100 calcium-binding protein A12 (S100A12) belongs to the S100 protein family and regulates amounts of inflammatory responses. S100A12 is significantly upregulated in acute KD and declined after IVIG treatment [53, 63,64,65], in accordance with aforementioned results. While in patients with giant CAAs, plasma S100A12 levels remain elevated [53, 63]. Besides, S100A12 could promote vasculitis by stimulating the production of IL-1β, which directly induces CA endothelial cell dysfunction [64] and impacts the risk of the formation of CAL in children with KD [65].
The infiltration profile of 22 immune cells in KD and HC was assessed using CIBERSORTx. Gamma delta T cells, monocytes, M0 macrophages, activated dendritic cells, activated mast cells and neutrophils were significantly augmented in KDs, which may explain the contribution of innate immunity to the occurrence of vasculitis in KD [66,67,68]. The fractions of memory B cells, plasma cells, CD8+ T cells, CD4+ naïve T cells, CD4+ resting memory T cells, CD4+ activated memory T cells, resting NK cells, activated NK cells, M2 macrophages, resting dendritic cells and resting mast cells were decreased in KDs, which has been demonstrated in several studies [13, 69,70,71,72]. Furthermore, by performing correlation analysis between node genes and immune cells, we discovered that all the node genes have correlation with neutrophils, which has been demonstrated in previous studies [73,74,75,76,77]. Therefore, we can infer that the five-gene signature is involved in the process of immune cell-mediated occurrence of KD, and our study provides a reference for the study of immune mechanisms in the pathogenesis of KD.
Target miRNAs and the target lncRNAs of these miRNAs were predicted for the node genes, which may reveal the mechanism through which the node genes are adjusted at the transcriptome level. Hsa-miR-524-5p and hsa-miR-204-5p have been found to be considerably downregulated in patients with heart disease as compared to controls [78, 79]. LncRNA-NEAT1 is associated with NLRP3 inflammasome to augment their aggregation and can also promote the production of IL-1β [80, 81]. Besides, it could regulate the expression of chemokines and cytokines, such as IL-6 [82]. NORAD modulates inflammation and atherosclerosis in various cardiovascular diseases [83]. Furthermore, XIST is involved in the pathogenesis of SLE and rheumatoid arthritis (RA) [84] and can promote the expression of MMP3 and caspase-3 [85], which are important molecules contributing to KD [86, 87]. Finally, three intact ceRNA network(s) was constructed, namely, NEAT1/NORAD/XIST-hsa-miR-524-5p-ADM, NEAT1/NORAD/XIST-hsa-miR-204-5p-ALPL, NEAT1/NORAD/XIST-hsa-miR-524-5p/hsa-miR-204-5p-MMP9. We hypothesize that this ceRNA network has a pivotal role in KD.
However, the disadvantage of our study is obvious. Compared to RNA-seq technique, which sequences the entire transcriptome, the datasets employed in our study only provide the profiles of predefined transcripts or genes through hybridization which could not reveal the full picture of gene expression. Besides, our analysis is based on prediction, and the exact function of each molecule in the process of KD needs to be verified by further cell and animal experiments. In addition, a larger cohort is needed to investigate the unknown process underlying KD. And the clinical information of samples was not comprehensive, which may prevent us from discovering other influence factors.
Conclusion
In conclusion, we identified a five-gene signature with the aid of WGCNA, LASSO regression analysis and algorithms in Cytoscape, and ultimately acquired three integrated ceRNA network: NEAT1/NORAD/XIST-hsa-miR-524-5p-ADM, NEAT1/NORAD/XIST-hsa-miR-204-5p-ALPL, NEAT1/NORAD/XIST-hsa-miR-524-5p/hsa-miR-204-5p-MMP9, with online databases. Our bioinformatic analysis offers potential biomarkers for the early diagnosis of KD and might perfect the regulatory mechanism from the perspective of ceRNA networks.
Availability of data and materials
The following data were offered considering data availability:
The original microarray data is available at Gene Expression Omnibus: GSE68004 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68004),
GSE73461 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE73461),
GSE18606 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE18606),
GSE63881 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63881).
All data obtained in this paper are contained in the figures and tables (Figs. 1, 2, 3, 4, 5 and 6; Tables 1, 2, 3 and 4; Figures S1, S2 and S3; Table S1).
Abbreviations
- A:
-
Acute KD patients
- ADM:
-
Adrenomedullin
- ALPL:
-
Alkaline phosphatase
- AUC:
-
Area under the curve
- BP:
-
Biological process
- C:
-
Convalescent KD patients
- CA:
-
Coronary arteries
- CAA:
-
Coronary artery aneurysm
- CAL:
-
Coronary artery lesion
- ceRNAs:
-
Competing endogenous RNAs
- CRP:
-
C-reactive protein
- DEGs:
-
Differential expressed genes
- EPC:
-
Edge Percolated Component
- FC:
-
Fold change
- FDR:
-
False discovery rate
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene Ontology
- GS:
-
Gene significances
- HC:
-
Health controls
- HK3:
-
Hexokinase 3
- Ig:
-
Immunoglobulin
- IL-6:
-
Interleukin-6
- IVIG:
-
Intravenous immunoglobulin
- KD:
-
Kawasaki disease
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LASSO:
-
Least absolute shrinkage and selection operator
- MCC:
-
Maximal Clique Centrality
- MM:
-
Module memberships
- MMP-9:
-
Matrix metalloproteinase 9
- MNC:
-
Maximum Neighborhood Component
- MS:
-
Module significance
- ncRNAs:
-
Noncoding RNAs
- PCA:
-
Principal component analysis
- PPI:
-
Protein–protein interaction
- RA:
-
Rheumatoid arthritis
- ROC:
-
Receiver operating characteristic
- S100A12:
-
S100 calcium-binding protein A12
- SLE:
-
Systemic lupus erythematosus
- VSMC:
-
Vascular smooth muscle cell
- WGCNA:
-
Weighted gene correlation network analysis
References
Del Principe D, Pietraforte D, Gambardella L, Marchesi A. Tarissi de Jacobis I, Villani A, Malorni W, Straface E: Pathogenetic determinants in Kawasaki disease: the haematological point of view. J Cell Mol Med. 2017;21(4):632–9.
Tian J, Lv HT, An XJ, Ling N, Xu F. Endothelial microparticles induce vascular endothelial cell injury in children with Kawasaki disease. Eur Rev Med Pharmacol Sci. 2016;20(9):1814–8.
Newburger JW, Takahashi M, Beiser AS, Burns JC, Bastian J, Chung KJ, Colan SD, Duffy CE, Fulton DR, Glode MP, et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N Engl J Med. 1991;324(23):1633–9.
Kuwabara M, Yashiro M, Kotani K, Tsuboi S, Ae R, Nakamura Y, Yanagawa H, Kawasaki T. Cardiac lesions and initial laboratory data in Kawasaki disease: a nationwide survey in Japan. J Epidemiol. 2015;25(3):189–93.
Parthasarathy P, Agarwal A, Chawla K, Tofighi T, Mondal TK. Upcoming biomarkers for the diagnosis of Kawasaki disease: A review. Clin Biochem. 2015;48(16–17):1188–94.
Li Y, Zheng Q, Zou L, Wu J, Guo L, Teng L, Zheng R, Jung LKL, Lu M. Kawasaki disease shock syndrome: clinical characteristics and possible use of IL-6, IL-10 and IFN-γ as biomarkers for early recognition. Pediatr Rheumatol Online J. 2019;17(1):1.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Zhang Q, Li J, Xue H, Kong L, Wang Y. Network-based methods for identifying critical pathways of complex diseases: a survey. Mol Biosyst. 2016;12(4):1082–9.
Engebretsen S, Bohlin J. Statistical predictions with glmnet. Clin Epigenetics. 2019;11(1):123.
Wright VJ, Herberg JA, Kaforou M, Shimizu C, Eleftherohorinou H, Shailes H, Barendregt AM, Menikou S, Gormley S, Berk M, et al. Diagnosis of Kawasaki Disease Using a Minimal Whole-Blood Gene Expression Signature. JAMA Pediatr. 2018;172(10):e182293.
Ling XB, Lau K, Kanegaye JT, Pan Z, Peng S, Ji J, Liu G, Sato Y, Yu TT, Whitin JC, et al. A diagnostic algorithm combining clinical and molecular data distinguishes Kawasaki disease from other febrile illnesses. BMC Med. 2011;9:130.
Hoang LT, Shimizu C, Ling L, Naim AN, Khor CC, Tremoulet AH, Wright V, Levin M, Hibberd ML, Burns JC. Global gene expression profiling identifies new therapeutic targets in acute Kawasaki disease. Genome Med. 2014;6(11):541.
Chaudhary H, Nameirakpam J, Kumrah R, Pandiarajan V, Suri D, Rawat A, Singh S. Biomarkers for Kawasaki Disease: Clinical Utility and the Challenges Ahead. Frontiers Pediatr. 2019;7:242.
Kuniyoshi Y, Tokutake H, Takahashi N, Kamura A, Yasuda S, Tashiro M. Comparison of Machine Learning Models for Prediction of Initial Intravenous Immunoglobulin Resistance in Children With Kawasaki Disease. Frontiers Pediatr. 2020;8:570834.
Sleeper LA, Minich LL, McCrindle BM, Li JS, Mason W, Colan SD, Atz AM, Printz BF, Baker A, Vetter VL, et al. Evaluation of Kawasaki disease risk-scoring systems for intravenous immunoglobulin resistance. J Pediatr. 2011;158(5):831-835.e833.
Nie H, Wang S, Wu Q, Xue D, Zhou W. Five immune-gene-signatures participate in the development and pathogenesis of Kawasaki disease. Immun Inflamm Dis. 2021;9(1):157–66.
Chang D, Qian C, Li H, Feng H. Comprehensive analyses of DNA methylation and gene expression profiles of Kawasaki disease. J Cell Biochem. 2019;120(8):13001–11.
Esmaeili S, Mehrgou A, Kakavandi N, Rahmati Y. Exploring Kawasaki disease-specific hub genes revealing a striking similarity of expression profile to bacterial infections using weighted gene co-expression network analysis (WGCNA) and co-expression modules identification tool (CEMiTool): An integrated bioinformatics and experimental study. Immunobiology. 2020;225(4):151980.
Liu D, Song M, Jing F, Liu B, Yi Q. Diagnostic Value of Immune-Related Genes in Kawasaki Disease. Front Genet. 2021;12:763496.
Jaggi P, Mejias A, Xu Z, Yin H, Moore-Clingenpeel M, Smith B, Burns JC, Tremoulet AH, Jordan-Villegas A, Chaussabel D, et al. Whole blood transcriptional profiles as a prognostic tool in complete and incomplete Kawasaki Disease. PLoS ONE. 2018;13(5):e0197858.
Fury W, Tremoulet AH, Watson VE, Best BM, Shimizu C, Hamilton J, Kanegaye JT, Wei Y, Kao C, Mellis S, et al. Transcript abundance patterns in Kawasaki disease patients with intravenous immunoglobulin resistance. Hum Immunol. 2010;71(9):865–73.
Taminau J, Meganck S, Lazar C, Steenhoff D, Coletta A, Molter C, Duque R, de Schaetzen V, Weiss Solís DY, Bersini H, et al. Unlocking the potential of publicly available microarray data using inSilicoDb and inSilicoMerging R/Bioconductor packages. BMC Bioinformatics. 2012;13:335.
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics (Oxford, England). 2012;28(6):882–3.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Kolde R. Pretty Heatmaps. R package. 2015. https://cran.r-project.org/web/packages/pheatmap/index.html.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2022;51(D1):D587–92.
Ito K, Murphy D. Application of ggplot2 to Pharmacometric Graphics. CPT Pharmacometrics Syst Pharmacol. 2013;2(10):e79.
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561-568.
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11.
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Müller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7.
Mitteer DR, Greer BD. Erratum: Publisher Correction to: Using GraphPad Prism’s Heat Maps for Efcient, Fine-Grained Analyses of Single-Case Data. Behav Anal Pract. 2022;15(2):515.
R package “corrplot”: visualization of a correlation matrix. R package version 0.84. https://Github.Com/Taiyun/Corrplot.
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128.
Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic acids Res. 2014;42(66):D92-97.
Hara T, Nakashima Y, Sakai Y, Nishio H, Motomura Y, Yamasaki S. Kawasaki disease: a matter of innate immunity. Clin Exp Immunol. 2016;186(2):134–43.
Sundel RP. Kawasaki disease. Rheum Dis Clin North Am. 2015;41(1):63–73 (viii).
Wu J, Fang X, Xia X. Identification of Key Genes and Pathways associated with Endometriosis by Weighted Gene Co-expression Network Analysis. Int J Med Sci. 2021;18(15):3425–36.
Tian Z, He W, Tang J, Liao X, Yang Q, Wu Y, Wu G. Identification of Important Modules and Biomarkers in Breast Cancer Based on WGCNA. Onco Targets Ther. 2020;13:6805–17.
Li Y, Zhao J, Chen R, Chen S, Xu Y, Cai W. Integration of clinical and transcriptomics reveals programming of the lipid metabolism in gastric cancer. BMC Cancer. 2022;22(1):955.
Xie Y, Han B. Exploring the relationship between pyroptosis, infiltrating immune cells and Kawasaki disease with resistance to intravenous immunoglobulin (IVIG) via bioinformatic analysis. Immunobiology. 2022;227(5):152261.
Wu ZY, Shi ZY. The prognostic value and immune landscapes of m1A/m5C/m6A-associated lncRNA signature in osteosarcoma. Eur Rev Med Pharmacol Sci. 2022;26(16):5868–83.
Feng X, Zhang Y, Du M, Li S, Ding J, Wang J, Wang Y, Liu P. Identification of diagnostic biomarkers and therapeutic targets in peripheral immune landscape from coronary artery disease. J Trans Med. 2022;20(1):399.
Wang Z, Liu J, Li M, Lian L, Cui X, Ng TW, Zhu M. Integrated bioinformatics analysis uncovers characteristic genes and molecular subtyping system for endometriosis. Front Pharmacol. 2022;13:932526.
Li Z, Huang B, Yi W, Wang F, Wei S, Yan H, Qin P, Zou D, Wei R, Chen N. Identification of Potential Early Diagnostic Biomarkers of Sepsis. J Inflamm Res. 2021;14:621–31.
Lai Y, Lin C, Lin X, Wu L, Zhao Y, Shao T, Lin F. Comprehensive Analysis of Molecular Subtypes and Hub Genes of Sepsis by Gene Expression Profiles. Front Genet. 2022;13:884762.
Nishida K, Watanabe K, Echigo S, Mayumi M, Nishikimi T. Increased plasma adrenomedullin levels in Kawasaki disease with coronary artery involvement. Am J Med. 2001;111(2):165–6.
Nomura I, Abe J, Noma S, Saito H, Gao B, Wheeler G, Leung DY. Adrenomedullin is highly expressed in blood monocytes associated with acute Kawasaki disease: a microarray gene expression study. Pediatric Res. 2005;57(1):49–55.
Bunton DC, Petrie MC, Hillier C, Johnston F, McMurray JJ. The clinical relevance of adrenomedullin: a promising profile? Pharmacol Ther. 2004;103(3):179–201.
Wong HK, Cheung TT, Cheung BM. Adrenomedullin and cardiovascular diseases. JRSM Cardiovasc Dis. 2012;1(5):cvd2012-012003.
Grabowski P. Physiology of Bone. Endocr Dev. 2015;28:33–55.
Qian G, Xu L, Qin J, Huang H, Zhu L, Tang Y, Li X, Ma J, Ma Y, Ding Y, et al. Leukocyte proteomics coupled with serum metabolomics identifies novel biomarkers and abnormal amino acid metabolism in Kawasaki disease. J Proteomics. 2021;239:104183.
Cheung BM, Ong KL, Cheung RV, Wong LY, Wat NM, Tam S, Leung GM, Cheng CH, Woo J, Janus ED, et al. Association between plasma alkaline phosphatase and C-reactive protein in Hong Kong Chinese. Clin Chem Lab Med. 2008;46(4):523–7.
Kunutsor SK, Bakker SJ, Kootstra-Ros JE, Gansevoort RT, Gregson J, Dullaart RP. Serum Alkaline Phosphatase and Risk of Incident Cardiovascular Disease: Interrelationship with High Sensitivity C-Reactive Protein. PLoS ONE. 2015;10(7):e0132822.
Wang XK, Gao C, Zhong HQ, Kong XY, Qiao R, Zhang HC, Chen BY, Gao Y, Li B. TNAP-a potential cytokine in the cerebral inflammation in spastic cerebral palsy. Front Mol Neurosci. 2022;15:926791.
Tuo Z, Zheng X, Zong Y, Li J, Zou C, Lv Y, Liu J. HK3 is correlated with immune infiltrates and predicts response to immunotherapy in non-small cell lung cancer. Clin Transl Med. 2020;10(1):319–30.
Geng Z, Liu J, Hu J, Wang Y, Tao Y, Zheng F, Wang Y, Fu S, Wang W, Xie C, et al. Crucial transcripts predict response to initial immunoglobulin treatment in acute Kawasaki disease. Sci Rep. 2020;10(1):17860.
Senzaki H. The pathophysiology of coronary artery aneurysms in Kawasaki disease: role of matrix metalloproteinases. Arch Dis Child. 2006;91(10):847–51.
Shimizu C, Matsubara T, Onouchi Y, Jain S, Sun S, Nievergelt CM, Shike H, Brophy VH, Takegawa T, Furukawa S, et al. Matrix metalloproteinase haplotypes associated with coronary artery aneurysm formation in patients with Kawasaki disease. J Human Genet. 2010;55(12):779–84.
Korematsu S, Ohta Y, Tamai N, Takeguchi M, Goto C, Miyahara H, Kawano T, Izumi T. Cell distribution differences of matrix metalloproteinase-9 and tissue inhibitor of matrix metalloproteinase-1 in patients with Kawasaki disease. Pediatr Infect Dis J. 2012;31(9):973–4.
Johnson JL, Dwivedi A, Somerville M, George SJ, Newby AC. Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler Thromb Vasc Biol. 2011;31(9):e35-44.
Lech M, Guess J, Duffner J, Oyamada J, Shimizu C, Hoshino S, Farutin V, Bulik DA, Gutierrez B, Sarvaiya H, et al. Circulating Markers of Inflammation Persist in Children and Adults With Giant Aneurysms After Kawasaki Disease. Circ Genom Precis Med. 2019;12(4):e002433.
Armaroli G, Verweyen E, Pretzer C, Kessel K, Hirono K, Ichida F, Okabe M, Cabral DA, Foell D, Brown KL, et al. Monocyte-Derived Interleukin-1β As the Driver of S100A12-Induced Sterile Inflammatory Activation of Human Coronary Artery Endothelial Cells: Implications for the Pathogenesis of Kawasaki Disease. Arthritis Rheumatol. 2019;71(5):792–804.
Fu LY, Qiu X, Deng QL, Huang P, Pi L, Xu Y, Che D, Zhou H, Lu Z, Tan Y, et al. The IL-1B Gene Polymorphisms rs16944 and rs1143627 Contribute to an Increased Risk of Coronary Artery Lesions in Southern Chinese Children with Kawasaki Disease. J Immunol Res. 2019;2019:4730507.
Furukawa S, Matsubara T, Yabuta K. Mononuclear cell subsets and coronary artery lesions in Kawasaki disease. Arch Dis Child. 1992;67(6):706–8.
Takahashi K, Oharaseki T, Yokouchi Y, Hiruta N, Naoe S. Kawasaki disease as a systemic vasculitis in childhood. Ann Vasc Dis. 2010;3(3):173–81.
Ding Y, Li G, Xiong LJ, Yin W, Liu J, Liu F, Wang RG, Xia K, Zhang SL, Zhao L. Profiles of responses of immunological factors to different subtypes of Kawasaki disease. BMC Musculoskelet Disord. 2015;16:315.
Furuno K, Yuge T, Kusuhara K, Takada H, Nishio H, Khajoee V, Ohno T, Hara T. CD25+CD4+ regulatory T cells in patients with Kawasaki disease. J Pediatr. 2004;145(3):385–90.
Kuijpers TW, Wiegman A, van Lier RA, Roos MT, Wertheim-van Dillen PM, Pinedo S, Ottenkamp J. Kawasaki disease: a maturational defect in immune responsiveness. J Infect Dis. 1999;180(6):1869–77.
Kumrah R, Vignesh P, Rawat A, Singh S. Immunogenetics of Kawasaki disease. Clin Rev Allergy Immunol. 2020;59(1):122–39.
Hirata S, Nakamura Y, Yanagawa H. Incidence rate of recurrent Kawasaki disease and related risk factors: from the results of nationwide surveys of Kawasaki disease in Japan. Acta Paediatrica (Oslo, Norway : 1992). 2001;90(1):40–4.
Zheng PF, Chen LZ, Liu P, Pan HW, Fan WJ, Liu ZY. Identification of immune-related key genes in the peripheral blood of ischaemic stroke patients using a weighted gene coexpression network analysis and machine learning. J Transl Med. 2022;20(1):361.
He GD, Huang YQ, Liu L, Huang JY, Lo K, Yu YL, Chen CL, Zhang B, Feng YQ. Association of Circulating, Inflammatory-Response Exosomal mRNAs With Acute Myocardial Infarction. Front Cardiovasc Med. 2021;8:712061.
Qian H, Ge A, Jiang JJ, Xu JF. Necroptosis-related subtypes are associated with bronchiectasis in pulmonary non-tuberculous mycobacteria-infected patients: a perspective based on transcriptomic analysis. European journal of clinical microbiology & infectious diseases: official publication of the European Society of Clinical Microbiology. 2023;42(2):141–52.
Yoshido A, Sudo G, Takasawa A, Aoki H, Kitajima H, Yamamoto E, Niinuma T, Harada T, Kubo T, Sasaki H, et al. Serum amyloid A1 recruits neutrophils to the invasive front of T1 colorectal cancers. J Gastroenterol Hepatol. 2023;38(2):301–10.
Eddins DJ, Yang J, Kosters A, Giacalone V, Pechuan X, Chandler JD, Eum J, Babcock BR, Dobosh BS, Hernández MR, et al. Transcriptional reprogramming of infiltrating neutrophils drives lung disease in severe COVID-19 despite low viral load. Blood advances. 2022;2022008834.
Yamada R, Okumura S, Kono Y, Miyazaki A, Niwa Y, Ito T, Ueda S, Ishiguro T, Yoshinaga M, Fujiwara W, et al. Effect of cardiac rehabilitation on circulating microRNA expression in heart failure: a preliminary study. Fujita Med J. 2021;7(3):76–82.
Rong J, Pan H, He J, Zhang Y, Hu Y, Wang C, Fu Q, Fan W, Zou Q, Zhang L, et al. Long non-coding RNA KCNQ1OT1/microRNA-204-5p/LGALS3 axis regulates myocardial ischemia/reperfusion injury in mice. Cell Signal. 2020;66:109441.
Zhang P, Cao L, Zhou R, Yang X, Wu M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat Commun. 2019;10(1):1495.
Wang L, Xia JW, Ke ZP, Zhang BH. Blockade of NEAT1 represses inflammation response and lipid uptake via modulating miR-342-3p in human macrophages THP-1 cells. J Cell Physiol. 2019;234(4):5319–26.
Zhang F, Wu L, Qian J, Qu B, Xia S, La T, Wu Y, Ma J, Zeng J, Guo Q, et al. Identification of the long noncoding RNA NEAT1 as a novel inflammatory regulator acting through MAPK pathway in human lupus. J Autoimmun. 2016;75:96–104.
Zhang X, Kan X, Shen J, Li J. Increased long non-coding RNA NORAD reflects serious cardiovascular stenosis, aggravated inflammation status, and higher lipid level in coronary heart disease. J Clin Lab Anal. 2022;36(11):e24717.
Bost C, Arleevskaya MI, Brooks WH, Plaza S, Guery JC, Renaudineau Y. Long non-coding RNA Xist contribution in systemic lupus erythematosus and rheumatoid arthritis. Clin Immunol. 2022;236:108937.
He J, Yang J, Shen T, He J. Overexpression of long non-coding RNA XIST promotes IL-1β-induced degeneration of nucleus pulposus cells through targeting miR-499a-5p. Mol Cell Probes. 2021;57:101711.
Park JA, Shin KS, Kim YW. Polymorphism of matrix metalloproteinase-3 promoter gene as a risk factor for coronary artery lesions in Kawasaki disease. J Korean Med Sci. 2005;20(4):607–11.
Onouchi Y, Ozaki K, Buns JC, Shimizu C, Hamada H, Honda T, Terai M, Honda A, Takeuchi T, Shibuta S, et al. Common variants in CASP3 confer susceptibility to Kawasaki disease. Hum Mol Genet. 2010;19(14):2898–906.
Acknowledgments
We would like to thank American Journal Expert (AJE) and Dr. Han for English language editing and review services. And we also thank the contributors of the GEO databases for the availability of the data.
Ethical Statement
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This study was supported by the National Natural Science Foundation of China (no.81873498); Jinan Science and Technology Development Plan (after subsidy) (no.202134015); Special Expert of Taishan Scholars (no.201511099).
Author information
Authors and Affiliations
Contributions
1. YXX conceived and designed the methods, conducted the analysis, prepared figures and tables, edited the drafts and the final version of the paper and approved the final draft. 2. HSS conceived the methods, reviewed the drafts of the paper and approved the final draft. 3. BH reviewed the drafts of the paper and approved the final draft. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ethics Committee of Shandong Provincial Hospital Affiliated to Shandong University ruled that no formal ethics approval was required in this particular case. Access to the database is on request. All data were obtained by the GEO database (https://www.ncbi.nlm.nih.gov/geo/). All methods were performed in accordance with the Declaration of Helsinki for all human participants.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Soft threshold analysis is used to acquire the scale-free fit index of network topology.
Additional file 2: Figure S2.
Validation of hub genes. Principal component analyses (PCA) of the training set (A) and GSE18606 (B). Red circles represent KD, turquoise circles represent HC.
Additional file 3: Figure S3.
Correlation among five node genes and immune cells in the training set. The number and the color in the dots mean the strength of the correlation; red indicates the positive correlation, and blue represents the negative. And the asterisk in the dots means the p-value (-: non-significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001).
Additional file 4: Table S1.
The results of all the statistical tests. DEGs are the genes with p-values < 0.05 and |log2 FC| ≥ 2 between KD and HC. GENES_IN_MODULES presents the modular distribution of genes according to the clustering of the WGCNA analysis. ENRICHED_GO_OF_HUB_MODULE and ENRICHED_PATHWAYS_OF_HUB_MODULE are the results of the enrichment analysis of genes in hub module using “clusterprofiler” R package. CIBERSORTx presents the proportions of immune cells in KD and HC, and the p-values between them.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xie, Y., Shi, H. & Han, B. Bioinformatic analysis of underlying mechanisms of Kawasaki disease via Weighted Gene Correlation Network Analysis (WGCNA) and the Least Absolute Shrinkage and Selection Operator method (LASSO) regression model. BMC Pediatr 23, 90 (2023). https://doi.org/10.1186/s12887-023-03896-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-023-03896-4