
Abstract
Background
Colorectal cancer (CRC) is major cancer-related death. The aim of this study was to identify differentially expressed and differentially methylated genes, contributing to explore the molecular mechanism of CRC.
Methods
Firstly, the data of gene transcriptome and genome-wide DNA methylation expression were downloaded from the Gene Expression Omnibus database. Secondly, functional analysis of differentially expressed and differentially methylated genes was performed, followed by protein-protein interaction (PPI) analysis. Thirdly, the Cancer Genome Atlas (TCGA) dataset and in vitro experiment was used to validate the expression of selected differentially expressed and differentially methylated genes. Finally, diagnosis and prognosis analysis of selected differentially expressed and differentially methylated genes was performed.
Results
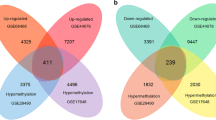
Up to 1958 differentially expressed (1025 up-regulated and 993 down-regulated) genes and 858 differentially methylated (800 hypermethylated and 58 hypomethylated) genes were identified. Interestingly, some genes, such as GFRA2 and MDFI, were differentially expressed-methylated genes. Purine metabolism (involved IMPDH1), cell adhesion molecules and PI3K-Akt signaling pathway were significantly enriched signaling pathways. GFRA2, FOXQ1, CDH3, CLDN1, SCGN, BEST4, CXCL12, CA7, SHMT2, TRIP13, MDFI and IMPDH1 had a diagnostic value for CRC. In addition, BEST4, SHMT2 and TRIP13 were significantly associated with patients’ survival.
Conclusions
The identified altered genes may be involved in tumorigenesis of CRC. In addition, BEST4, SHMT2 and TRIP13 may be considered as diagnosis and prognostic biomarkers for CRC patients.
Similar content being viewed by others

Background
Colorectal cancer (CRC) is major cancer-related death [1, 2]. Sustained cell proliferation and invasion, enhanced angiogenesis and metastasis, and drug resistance are the major characteristics of CRC [3, 4]. Various factors are related to the development of CRC, such as genetics, polyposis, chronic inflammation, inflammatory bowel disease, increased body mass index, little physical activity, cigarette smoking, alcohol abuse and particular dietary habits [5,6,7,8,9,10,11]. Clinically, main curative treatments for CRC are radiotherapy, chemotherapy and surgical removal of lesions. The survival outcome of CRC patients is worse, with a 5-year survival rate of only 14.0% [12]. Therefore, it is important to understand the pathological mechanism of CRC.
Simons CCJM et al. found that the CpG island methylated phenotype is a major factor contributing to CRC carcinogenesis [13]. Furthermore, gene expression regulation by aberrant DNA methylation is extensively described for CRC. For example, abnormal methylation of septin 9 (SEPT9) is frequently reported in CRC, and the SEPT9 methylation test has been used in early screening for CRC [14,15,16]. In order to further investigate the pathological mechanism of CRC, we performed both integrated analysis and DNA methylation analysis in the Gene Expression Omnibus database to find potential and valuable genes in CRC.
Methods
Datasets retrieval
We searched datasets from the GEO dataset with the keywords (Colorectal cancer) AND “Homo sapiens”[porgn:__txid9606]. All selected datasets were gene transcriptome and genome-wide DNA methylation expression data in the CRC tumor tissues and normal controls. Finally, a total of 3 datasets of gene transcriptome data (GSE113513, GSE87211 and GSE89076) and 2 datasets of genome-wide DNA methylation expression data (GSE101764 and GSE129364) were identified (Table 1). Clinical information of above datasets is shown in supplementary Table 1.
Identification of differentially expressed and differentially methylated genes
Firstly, scale standardization was carried out for the common genes in 3 datasets of gene transcriptome data. The metaMA and limma packages were used to identify differentially expressed genes [17]. P values and effect sizes from data were calculated either from classical or moderated t-tests. These p values were combined by the inverse normal method. Benjamini hochberg threshold was used to calculate the false discovery rate (FDR). Finally, differentially expressed genes were obtained with the criterion of FDR and |Combined.effect size| ≥ 1.5. In addition, quantile standardization was performed for the common genes in 2 datasets of genome-wide DNA methylation expression data. Benjamini hochberg threshold was used to calculate the FDR. COHCAP package in R language was used to identify differentially methylated genes under the threshold of |Δβ| > 0.3 and FDR < 0.05.
Functional analysis of differentially expressed and differentially methylated genes
To understand the function of differentially expressed and differentially methylated genes, we conducted Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis through David 6.8 (https://david.ncifcrf.gov/). FDR < 0.05 was considered as significant.
PPI network
The BioGRID database was used to retrieve the predicted interactions between top 50 proteins and other proteins. In the network, node and edge represents protein and the interactions, respectively.
Electronic and in vitro validation of differentially expressed and differentially methylated genes
The Cancer Genome Atlas (TCGA) dataset (involved 478 patients with CRC and 41 normal controls) was used to validate the expression of differentially expressed and differentially methylated genes. The expression result of these genes was shown by box plots.
In vitro validation QRT-PCR was also performed. The inclusion criteria of CRC patients was as follows: (1) Patients were diagnosed with CRC according to the pathological examination; (2) Patients underwent radical resection of CRC for the first time and received no chemoradiotherapy before; (3) patients had complete clinical data including medical history of present illness, personal history, family history, detailed physical examination data and postoperative pathological data. The exclusion criteria of CRC patients were as follows: (1) patients had other colorectal tumors, carcinoid, malignant melanoma, malignant lymphoma and so on; (2) patients had multiple primary CRC, familial adenomatous polyposis and concurrent or previous malignancy. According to the above criteria, 5 CRC patients were enrolled. Clinical information of these CRC patients was listed in Table 2. The tumor tissue and para-carcinoma tissue of these patients was collected. All participating individuals provided informed consent with the approval of the ethics committee of the local hospital. All the experimental protocol for involving humans was in accordance to guidelines of national/international/institutional or Declaration of Helsinki.
Total RNA of the tissue and para-carcinoma tissue was extracted and synthesized DNA by FastQuant cDNA first strand synthesis kit (TIANGEN). Then real-time PCR was performed in the SuperReal PreMix Plus (SYBR Green) (TIANGEN). ACTB and GAPDH were used for internal reference. Relative mRNAs expression was analyzed by log2 (fold change) method.
Diagnosis and prognosis analysis of differentially expressed and differentially methylated genes
We performed the ROC and survival analysis to assess the diagnostic and prognostic value of differentially expressed and differentially methylated genes in the TCGA dataset.
Results
Differentially expressed and differentially methylated genes in the GEO dataset
There were 17,323 common genes in 3 datasets of gene transcriptome data. After scale standardization and differential expression analysis, a total of 1958 differentially expressed genes were identified in CRC. Top 20 differentially expressed genes were listed in Table 3. The heat map of top 100 differentially expressed genes was shown in Fig. 1. Additionally, there were 485,511 common methylation sites in 2 datasets of genome-wide DNA methylation expression data. After quantile standardization and differential methylation analysis, a total of 2661 differentially methylated sites were screened out in CRC. Correspondingly, there were 858 differentially methylated genes (800 hypermethylated genes and 58 hypomethylated genes) in these differentially methylated sites. The Manhattan and heat map of all differential methylated sites was shown in Fig. 2 and Fig. 3, respectively. Some differentially expressed genes, such as down-regulated GFRA2 was hypermethylated gene. Up-regulated MDFI was hypomethylated gene.

The heat map of top 100 differentially expressed genes in CRC. Diagram presents the result of a two-way hierarchical clustering of top 100 differentially expressed genes and samples. Each row and each column represents a differentially expressed gene and a sample, respectively

The Manhattan of all differential methylation sites in CRC. The x-axis represents the chromosome, the y-axis represents the -log10 (FDR) of differential methylation sites

The heat map of all differentially methylated sites in CRC. Diagram presents the result of a two-way hierarchical clustering of all differentially methylated sites and samples. Each row and each column represents a differentially methylated site and a sample, respectively
Biological function of differentially expressed and differentially methylated genes
All differentially expressed genes were the most significantly enriched in the biological process of DNA replication (Fig. 4A), cytological component of nucleoplasm (Fig. 4B) and molecular function of protein binding (Fig. 4C). In addition, cell cycle, DNA replication and purine metabolism (involved IMPDH1) were the most remarkably enriched signaling pathways of differentially expressed genes (Table 4).

A Top 15 significantly enriched biological processes of differentially expressed genes. The x-axis and y-axis represents the count of differentially expressed genes and terms of biological process, respectively. B Top 15 significantly enriched cytological components of differentially expressed genes. The x-axis and y-axis represents the count of differentially expressed genes and terms of cytological component, respectively. C Top 15 significantly enriched molecular functions of differentially expressed genes. The x-axis and y-axis represents the count of differentially expressed genes and terms of molecular function, respectively
Additionally, all differentially methylated genes were the most significantly enriched in the biological process of homophilic cell adhesion via plasma membrane adhesion molecules (Fig. 5A), cytological component of plasma membrane (Fig. 5B) and molecular function of sequence-specific DNA binding (Fig. 5C). Neuroactive ligand-receptor interaction, calcium signaling pathway, cAMP signaling pathway, cell adhesion molecules (CAMs), PI3K-Akt and Rap1 were the most remarkably enriched KEGG signaling pathways of all differentially methylated genes (Fig. 5D).

A Top 10 significantly enriched biological processes of differentially methylated genes. The x-axis and y-axis represents the count of differentially methylated genes and terms of biological process, respectively. B Top 10 significantly enriched cytological components of differentially methylated genes. The x-axis and y-axis represents the count of differentially methylated genes and terms of cytological component, respectively. C Top 10 significantly enriched molecular functions of differentially methylated genes. The x-axis and y-axis represents the count of differentially methylated genes and terms of molecular function, respectively. D Top 6 significantly enriched KEGG signaling pathways of differentially methylated genes. The x-axis and y-axis represents the count of differentially methylated genes and KEGG terms, respectively. The KEGG source has been obtained the permission from the Kanehisa laboratories (www.kegg.jp/feedback/copyright.html)
PPI network
PPI networks of top 100 differentially expressed genes were shown in Fig. 6. The top 10 proteins with a high degree (interaction with other proteins) were SHMT2 (degree = 44, up-regulation), FOXQ1 (degree = 19, up-regulation), TRIP13 (degree = 17, up-regulation), MDFI (degree = 16, up-regulation), CSE1L (degree = 11, up-regulation), DPEP1 (degree = 7, up-regulation), CPNE7 (degree = 7, up-regulation), IMPDH1 (degree = 7, up-regulation), UBE2C (degree = 6, up-regulation) and SLC7A5 (degree = 6, up-regulation).

PPI networks. The red and green colors represent up-regulated and down-regulated genes, respectively. Circular with black border represent top 10 up-regulated and down-regulated genes, respectively
Expression validation of differentially expressed and differentially methylated genes
The TCGA dataset was firstly used to validate the expression of GFRA2, FOXQ1, CDH3, CLDN1, SCGN, BEST4, CXCL12, CA7, SHMT2, TRIP13, MDFI and IMPDH1 (Fig. 7). The expression of FOXQ1, CDH3, CLDN1, SHMT2, TRIP13, MDFI and IMPDH1 was up-regulated, while GFRA2, SCGN, BEST4, CXCL12 and CA7 were down-regulated in CRC. The in vitro experiment was applied to further validate the expression of GFRA2, FOXQ1, CDH3, CLDN1, SCGN, BEST4 and CXCL12 in 5 patients. The expression of FOXQ1, CDH3 and CLDN1 was significantly up-regulated, while the expression of GFRA2, SCGN, BEST4 and CXCL12 was remarkably down-regulated in CRC (Fig. 8). All the validation result was in line with the bioinformatics analysis.

Expression box plots of GFRA2, FOXQ1, CDH3, CLDN1, SCGN, BEST4, CXCL12, CA7, SHMT2, TRIP13, MDFI and IMPDH1 in the TCGA dataset

The in vitro QRT-PCR validation of GFRA2, FOXQ1, CDH3, CLDN1, SCGN, BEST4 and CXCL12 in CRC. Log2 (fold change) > 1 and log2 (fold change) < 1 represents up-regulation and down-regulation, respectively. *p < 0.05; **p < 0.01; ***p < 0.001
Diagnosis and survival prediction of key differentially expressed and differentially methylated genes
Firstly, we performed ROC curve analyses to assess the diagnosis ability of GFRA2, FOXQ1, CDH3, CLDN1, SCGN, BEST4, CXCL12, CA7, SHMT2, TRIP13, MDFI and IMPDH1 in the TCGA dataset (Fig. 9). The AUC of these genes was more than 0.7, which suggested that they had a diagnostic value for CRC. In addition, we further analyzed the potential prognostic value of these genes. The result showed that BEST4, SHMT2 and TRIP13 were considered to be remarkably negatively associated with survival (p < 0.05) time with CRC patients. The survival curves of GFRA2, FOXQ1, CDH3, CLDN1, SCGN, BEST4, CXCL12, CA7, SHMT2, TRIP13, MDFI and IMPDH1 were illustrated in Fig. 10.

The ROC curves of GFRA2, FOXQ1, CDH3, CLDN1, SCGN, BEST4, CXCL12, CA7, SHMT2, TRIP13, MDFI and IMPDH1 between CRC and normal controls. The ROC curves were used to show the diagnostic ability of these genes with 1-specificity and sensitivity

The survival curves of GFRA2, FOXQ1, CDH3, CLDN1, SCGN, BEST4, CXCL12, CA7, SHMT2, TRIP13, MDFI and IMPDH1 in CRC
Discussion
GDNF family receptor alpha 2 (GFRA2) plays an important role in immune cells and intermediate monocytes in cancer [18, 19]. It is reported that ret. proto-oncogene (Ret) signaling through the combination of GFRA2 and neurturin (NRTN) is associated with the development of enteric nervous system [20]. Macartney-Coxson DP et al. found that GFRA2 was remarkably down-regulated in the process of CRC and possibly related to liver metastasis [21]. In mice, the function inhibition of MyoD family inhibitor (MDFI) promotes the regeneration of the gastrocnemius muscle after injury [22]. In addition, MDFI is over expressed in CRC tumors and high expression of MDFI is associated with tumor metastasis [22]. In this study, we found that down-regulated GFRA2 and up-regulated MDFI were differentially expressed-methylated genes in CRC. This indicated that gene methylaton may be associated with gene expression changes. Moreover, GFRA2 and MDFI had a diagnostic value for CRC patients. Our study further demonstrated the key roles of GFRA2 and MDFI in the process of CRC.
Forkhead box Q1 (FOXQ1), a transcription factor, activates target mRNA expression to regulate CRC cell migration, growth, epithelial-mesenchymal transition and chemoresistance [23, 24]. It is found that FOXQ1 is over expressed in tumor tissues of CRC and its high expression is significantly related to the stage and lymph node metastasis of CRC [25]. In addition, knock-down of FOXQ1 gene reduces the activity of Wnt signaling pathway [25]. These reports suggest that FOXQ1 can be considered as a potential therapeutic target for CRC. Cadherin 3 (CDH3), involved in cell–cell adhesion, is used to detect lymph nodes metastatic in patients with CRC [26, 27]. It has been demonstrated that hypomethylation is associated with CRC [28]. Furthermore, CDH3 is more frequently demethylated in advanced CRC [29]. In CRC, silencing the CDH3 genes lead to a remarkable decrease in tumor cell viability and proliferation [30]. Claudin 1 (CLDN1) is associated with CRC tumor invasion, lymph node metastasis and tumor grade and stage [31]. High expression of CLDN1 has been found in primary and metastatic CRC, and CRC cell lines [32,33,34,35]. Additionally, CLDN1 is remarkably hypomethylated in tumor samples of CRC [31]. CLDN1 targeting with the anti-CLDN1 monoclonal antibody reduces growth and survival of CRC cells, which suggest that CLDN1 can be a potential new therapeutic target for CRC [36]. Herein, we found that expression FOXQ1, CDH3 and CLDN1 were top 10 up-regulated genes in CRC. Furthermore, FOXQ1, CDH3 and CLDN1 had a diagnostic value for CRC patients. Our findings may provide new insight into the cancer biology of CRC.
Secretagogin, EF-hand calcium binding protein (SCGN) expresses in normal endocrine tissues, such as neuroendocrine cells of gastrointestinal tract [37]. In mice, Scgn gene deficient leads to colitis, which highlights the role of Scgn in intestinal immune homeostasis [38]. The expression of bestrophin 4 (BEST4) is decreased in colon tumor, colon adenocarcinoma and rectal adenocarcinoma and CRC [39,40,41,42]. In addition, BEST4 expression is remarkably negatively related to the survival probability of patients with CRC after surgery [42]. C-X-C motif chemokine ligand 12 (CXCL12) plays important roles in the immune system. CXCL12 is associated with promotes CRC tumor cell growth, liver migration, survival rate and recurrence rate [43, 44]. It is reported that the CXCL12 gene polymorphism could contribute to CRC by mediating tumor angiogenesis, progression, metastasis and leukocyte migration [45]. It is assumed that the CXCL12-G801A polymorphism can be used to indicate and detect stage T2 CRC [46]. In addition, activation of the CXCL12/C-X-C motif chemokine receptor 4 (CXCR4) axis renders CRC cell less sensitive to radiotherapy [47]. Carbonic anhydrase 7 (CA7) is expressed in various normal tissues including colon [48]. Decreased expression of CA7 has been found in rectal cancer, rectal adenocarcinoma and CRC [49,50,51]. It is worth mentioning that CRC patients with lower CA7 expression had a remarkable shorter disease-specific survival in early stage tumors [51]. In the present study, we found that SCGN, BEST4, CXCL12 and CA7 were top 10 down-regulated genes in CRC. Both of them had a diagnostic value for patients with CRC. Interestingly, BEST4 was significantly related to survival time of CRC patients. Our result indicated that SCGN, BEST4, CXCL12 and CA7 could be involved in the development of CRC.
According to the PPI analysis, we found several high degree proteins encoded by differentially expressed genes, such as serine hydroxymethyltransferase 2 (SHMT2) and thyroid hormone receptor interactor 13 (TRIP13). SHMT2, a key regulator in the serine/glycine metabolism pathway, is involved in cancer proliferation [52, 53]. It is revealed that SHMT2 is up-regulated in colon cancer [54]. It is noted that SHMT2 is associated with the occurrence and development of CRC [55]. Moreover, SHMT2 regulation by acetylation plays a crucial role in colorectal carcinogenesis [56]. TRIP13 promotes CRC cell growth, proliferation, invasion, migration and subcutaneous tumor formation [57]. It is found that high expression of TRIP13 is related to poor prognosis in CRC [57]. Additionally, TRIP13 is involved in colorectal adenoma-to-carcinoma progression [58]. In our study, the expression of SHMT2 and TRIP13 was increased in CRC. Significantly, both SHMT2 and TRIP13 had a remarkable diagnostic and prognostic value for CRC.
In addition, we found some significantly enriched signaling pathways of identified genes, including purine metabolism (involved up-regulated inosine monophosphate dehydrogenase 1, IMPDH1), cell adhesion molecules and PI3K-Akt signaling pathway. Spurr IB et al. found that the targeting of de novo purine metabolism was a viable strategy to block tumor growth in dividing cancer cells [59]. It has been demonstrated that purine metabolism is associated with the tumorigenesis of CRC [60]. The over expression of IMPDH1 has been found in CRC [61]. Some cell adhesion molecules such as selectins and immunoglobulin superfamily proteins play necessary roles in the CRC metastasis [62]. Ngan CY and Zlobec I et al. found that some cell adhesion molecules including E-cadherin and CD44v6 were lost at the invasive front of CRC [63, 64]. The PI3K/Akt signaling pathway plays an important role in CRC and inhibition of the pathway is a potential therapeutic strategy of CRC [65, 66].
Conclusions
In summary, we have obtained numerous differentially expressed and differentially methylated genes in CRC. Among which, GFRA2 and MDFI, were differentially expressed-methylated genes. It is suggested that DNA methylation may affect the expression changes of gene. Interestingly, GFRA2, FOXQ1, CDH3, CLDN1, SCGN, BEST4, CXCL12, CA7, SHMT2, TRIP13, MDFI and IMPDH1 were considered as the potential diagnostic biomarkers for CRC. In addition, BEST4, SHMT2 and TRIP13 could be used for prognostic detection molecule in CRC patients. However, there are limitations to our study. Firstly, the larger numbers of samples are further needed; Secondly, pyrosequencing and the QRT-PCR of gene methylation are further needed to respectively validate the methylation status and investigate the expression changes of methylated genes. Thirdly, the deeper mechanism study of the CRC is also explored.
Availability of data and materials
All data generated or analysed during this study are publicly available from GSE113513 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113513), GSE87211 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE87211), GSE89076 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE89076), GSE101764 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101764) and GSE129364 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE129364).
Abbreviations
- BEST4:
-
Bestrophin 4
- CDH3:
-
Cadherin 3
- CA7:
-
Carbonic anhydrase 7
- CAMs:
-
Cell adhesion molecules
- CLDN1:
-
Claudin 1
- CRC:
-
Colorectal cancer
- CXCL12:
-
C-X-C motif chemokine ligand 12
- CXCR4:
-
CXCL12/C-X-C motif chemokine receptor 4
- FDR:
-
False discovery rate
- FOXQ1:
-
Forkhead box Q1
- GFRA2:
-
GDNF family receptor alpha 2
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MDFI:
-
MyoD family inhibitor
- NRTN:
-
Neurturin
- PPI:
-
Protein-protein interaction
- SCGN:
-
Secretagogin, EF-hand calcium binding protein
- SEPT9:
-
Septin 9
- SHMT2:
-
Serine hydroxymethyltransferase 2
- TCGA:
-
The Cancer Genome Atlas
- TRIP13:
-
Thyroid hormone receptor interactor 13
References
Christensen J, Bentz S, Sengstag T, Shastri VP, Anderle P. FOXQ1, a novel target of the Wnt pathway and a new marker for activation of Wnt signaling in solid tumors. PLoS One. 2013;8(3):e60051.
Yee YK, Tan VP, Chan P, Hung IF, Pang R, Wong BC. Epidemiology of colorectal cancer in Asia. J Gastroenterol Hepatol. 2009;24(12):1810–6.
Ellis LM. Angiogenesis and its role in colorectal tumor and metastasis formation. Semin Oncol. 2004;31(6 Suppl 17):3–9.
Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med. 2009;361(25):2449–60.
Cereda M, Gambardella G, Benedetti L, Iannelli F, Patel D, Basso G, et al. Patients with genetically heterogeneous synchronous colorectal cancer carry rare damaging germline mutations in immune-related genes. Nat Commun. 2016;7:12072.
IJspeert R. Clinical risk factors of colorectal cancer in patients with serrated polyposis syndrome: a multicentre cohort analysis. Gut. 2015.
Rogler G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014;345(2):235–41.
Johnson CM, Wei C, Ensor JE, Smolenski DJ, Amos CI, Levin B, et al. Meta-analyses of colorectal cancer risk factors. Cancer Causes Control. 2013;24(6):1207–22.
Watson AJ, Collins PD. Colon cancer: a civilization disorder. Digestive diseases (Basel, Switzerland). 2011;29(2):222–8.
Terry P, Giovannucci E, Michels KB, Bergkvist L, Hansen H, Holmberg L, et al. Fruit, vegetables, dietary fiber, and risk of colorectal cancer. J Natl Cancer Inst. 2001;93(7):525–33.
Michels KB, Edward G, Joshipura KJ, Rosner BA, Stampfer MJ, Fuchs CS, et al. Prospective study of fruit and vegetable consumption and incidence of colon and rectal cancers. J Natl Cancer Inst. 2000;92(21):1740–52.
Provenzale D, Gupta S, Ahnen DJ, Markowitz AJ, Chung DC, Mayer RJ, et al. NCCN guidelines insights: colorectal Cancer screening, version 1.2018. J Natl Compr Cancer Netw. 2018;16(8):939–49.
Simons CC, Hughes LA, Smits KM, Khalid-de Bakker CA, de Bruïne AP, Carvalho B, et al. A novel classification of colorectal tumors based on microsatellite instability, the CpG island methylator phenotype and chromosomal instability: implications for prognosis. Ann Oncol. 2013;24(8):2048–56.
Song L, Li Y. SEPT9: a specific circulating biomarker for colorectal Cancer. Adv Clin Chem. 2015;72:171–204.
Church TR, Wandell M, Lofton-Day C, Mongin SJ, Burger M, Payne SR, et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut. 2014;63(2):317–25.
Lamb YN, Dhillon S. Epi proColon(®) 2.0 CE: a blood-based screening test for colorectal Cancer. Mol Diagn Ther. 2017;21(2):225–32.
Marot G, Foulley JL, Mayer CD, Jaffrézic F. Moderated effect size and P-value combinations for microarray meta-analyses. Bioinformatics (Oxford, England). 2009;25(20):2692–9.
Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116(16):e74–80.
Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. 2011;118(5):e16–31.
Musser MA, Michelle Southard-Smith E. Balancing on the crest - evidence for disruption of the enteric ganglia via inappropriate lineage segregation and consequences for gastrointestinal function. Dev Biol. 2013;382(1):356–64.
Macartney-Coxson DP, Hood KA, Shi HJ, Ward T, Wiles A, O'Connor R, et al. Metastatic susceptibility locus, an 8p hot-spot for tumour progression disrupted in colorectal liver metastases: 13 candidate genes examined at the DNA, mRNA and protein level. BMC Cancer. 2008;8:187.
Sui Y, Li X, Oh S, Zhang B, Freeman WM, Shin S, et al. Opposite roles of the JMJD1A interaction partners MDFI and MDFIC in colorectal Cancer. Sci Rep. 2020;10(1):8710.
Kaneda H, Arao T, Tanaka K, Tamura D, Aomatsu K, Kudo K, et al. FOXQ1 is overexpressed in colorectal cancer and enhances tumorigenicity and tumor growth. Cancer Res. 2010;70(5):2053–63.
Qiao Y, Jiang X, Lee ST, Karuturi RK, Hooi SC, Yu Q. FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res. 2011;71(8):3076–86.
Peng X, Luo Z, Kang Q, Deng D, Wang Q, Peng H, et al. FOXQ1 mediates the crosstalk between TGF-β and Wnt signaling pathways in the progression of colorectal cancer. Cancer Biol Ther. 2015;16(7):1099–109.
Chen G, Han N, Li G, Li X, Li G, Li Z, et al. Time course analysis based on gene expression profile and identification of target molecules for colorectal cancer. Cancer Cell Int. 2016;16:22.
Shrout J, Yousefzadeh M, Dodd A, Kirven K, Blum C, Graham A, et al. Beta (2) microglobulin mRNA expression levels are prognostic for lymph node metastasis in colorectal cancer patients. Br J Cancer. 2008;98(12):1999–2005.
Schnekenburger M, Diederich M. Epigenetics offer new horizons for colorectal Cancer prevention. Curr Colorectal Cancer Rep. 2012;8(1):66–81.
Hibi K, Goto T, Mizukami H, Kitamura YH, Sakuraba K, Sakata M, et al. Demethylation of the CDH3 gene is frequently detected in advanced colorectal cancer. Anticancer Res. 2009;29(6):2215–7.
Broussard EK, Kim R, Wiley JC, Marquez JP, Annis JE, Pritchard D, et al. Identification of putative immunologic targets for colon cancer prevention based on conserved gene upregulation from preinvasive to malignant lesions. Cancer Prev Res (Philadelphia, Pa). 2013;6(7):666–74.
Hahn-Strömberg V, Askari S, Ahmad A, Befekadu R, Nilsson TK. Expression of claudin 1, claudin 4, and claudin 7 in colorectal cancer and its relation with CLDN DNA methylation patterns. Tumour Biol. 2017;39(4):1010428317697569.
Gröne J, Weber B, Staub E, Heinze M, Klaman I, Pilarsky C, et al. Differential expression of genes encoding tight junction proteins in colorectal cancer: frequent dysregulation of claudin-1, −8 and −12. Int J Color Dis. 2007;22(6):651–9.
Huo Q, Kinugasa T, Wang L, Huang J, Zhao J, Shibaguchi H, et al. Claudin-1 protein is a major factor involved in the tumorigenesis of colorectal cancer. Anticancer Res. 2009;29(3):851–7.
Miwa N, Furuse M, Tsukita S, Niikawa N, Nakamura Y, Furukawa Y. Involvement of claudin-1 in the beta-catenin/Tcf signaling pathway and its frequent upregulation in human colorectal cancers. Oncol Res. 2001;12(11–12):469–76.
Dhawan P, Singh AB, Deane NG, No Y, Shiou SR, Schmidt C, et al. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J Clin Invest. 2005;115(7):1765–76.
Cherradi S, Ayrolles-Torro A, Vezzo-Vié N, Gueguinou N, Denis V, Combes E, et al. Antibody targeting of claudin-1 as a potential colorectal cancer therapy. J Exp Clin Cancer Res. 2017;36(1):89.
Wagner L, Oliyarnyk O, Gartner W, Nowotny P, Groeger M, Kaserer K, et al. Cloning and expression of Secretagogin, a novel neuroendocrine- and pancreatic islet of Langerhans-specific Ca2+−binding protein. J Biol Chem. 2000;275(32):24740.
Sifuentes-Dominguez LF, Li H, Llano E, Liu Z, Singla A, Patel AS, et al. SCGN deficiency results in colitis susceptibility. Elife. 2019;8:e49910.
Mojica W, Hawthorn L. Normal colon epithelium: a dataset for the analysis of gene expression and alternative splicing events in colon disease. BMC Genomics. 2010;11:5.
Poursheikhani A, Abbaszadegan MR, Nokhandani N, Kerachian MA. Integration analysis of long non-coding RNA (lncRNA) role in tumorigenesis of colon adenocarcinoma. BMC Med Genet. 2020;13(1):108.
Zhu X, Wang D. Screening key lncRNAs for human rectal adenocarcinoma based on lncRNA-mRNA functional synergistic network. Cancer Med. 2019;8(8):3875–91.
Pan F, Chen T, Sun X, Li K, Jiang X, Försti A, et al. Prognosis prediction of colorectal Cancer using gene expression profiles. Front Oncol. 2019;9:252.
Kollmar O, Rupertus K, Scheuer C, Junker B, Tilton B, Schilling MK, et al. Stromal cell-derived factor-1 promotes cell migration and tumor growth of colorectal metastasis. Neoplasia (New York, NY). 2007;9(10):862–70.
Lin CH, Shih CH, Tseng CC, Yu CC, Tsai YJ, Bien MY, et al. CXCL12 induces connective tissue growth factor expression in human lung fibroblasts through the Rac1/ERK, JNK, and AP-1 pathways. PLoS One. 2014;9(8):e104746.
Dimberg J, Hugander A, Löfgren S, Wågsäter D. Polymorphism and circulating levels of the chemokine CXCL12 in colorectal cancer patients. Int J Mol Med. 2007;19(1):11–5.
Shi MD, Chen JH, Sung HT, Lee JS, Tsai LY, Lin HH. CXCL12-G801A polymorphism modulates risk of colorectal cancer in Taiwan. Arch Med Sci. 2013;9(6):999–1005.
Wang D, Jiao C, Zhu Y, Liang D, Zao M, Meng X, et al. Activation of CXCL12/CXCR4 renders colorectal cancer cells less sensitive to radiotherapy via up-regulating the expression of survivin. Exp Biol Med (Maywood, NJ). 2017;242(4):429–35.
Bootorabi F, Jänis J, Smith E, Waheed A, Kukkurainen S, Hytönen V, et al. Analysis of a shortened form of human carbonic anhydrase VII expressed in vitro compared to the full-length enzyme. Biochimie. 2010;92(8):1072–80.
Zhang ZY, Yao QZ, Liu HY, Guo QN, Qiu PJ, Chen JP, et al. Metabolic reprogramming-associated genes predict overall survival for rectal cancer. J Cell Mol Med. 2020;24(10):5842–9.
Hua Y, Ma X, Liu X, Yuan X, Qin H, Zhang X. Abnormal expression of mRNA, microRNA alteration and aberrant DNA methylation patterns in rectal adenocarcinoma. PLoS One. 2017;12(3):e0174461.
Yang GZ, Hu L, Cai J, Chen HY, Zhang Y, Feng D, et al. Prognostic value of carbonic anhydrase VII expression in colorectal carcinoma. BMC Cancer. 2015;15:209.
Yang X, Wang Z, Li X, Liu B, Liu M, Liu L, et al. SHMT2 Desuccinylation by SIRT5 drives Cancer cell proliferation. Cancer Res. 2018;78(2):372–86.
Kim SK, Jung WH, Koo JS. Differential expression of enzymes associated with serine/glycine metabolism in different breast cancer subtypes. PLoS One. 2014;9(6):e101004.
Lin C, Zhang Y, Chen Y, Bai Y, Zhang Y. Long noncoding RNA LINC01234 promotes serine hydroxymethyltransferase 2 expression and proliferation by competitively binding miR-642a-5p in colon cancer. Cell Death Dis. 2019;10(2):137.
He Z, Wang X, Zhang H, Liang B, Zhang J, Zhang Z, et al. High expression of folate cycle enzyme MTHFD1L correlates with poor prognosis and increased proliferation and migration in colorectal cancer. J Cancer. 2020;11(14):4213–21.
Wei Z, Song J, Wang G, Cui X, Zheng J, Tang Y, et al. Deacetylation of serine hydroxymethyl-transferase 2 by SIRT3 promotes colorectal carcinogenesis. Nat Commun. 2018;9(1):4468.
Sheng N, Yan L, Wu K, You W, Gong J, Hu L, et al. TRIP13 promotes tumor growth and is associated with poor prognosis in colorectal cancer. Cell Death Dis. 2018;9(3):402.
Sillars-Hardebol AH, Carvalho B, de Wit M, Postma C, Delis-van Diemen PM, Mongera S, et al. Identification of key genes for carcinogenic pathways associated with colorectal adenoma-to-carcinoma progression. Tumour Biol. 2010;31(2):89–96.
Spurr IB, Birts CN, Cuda F, Benkovic SJ, Blaydes JP, Tavassoli A. Targeting tumour proliferation with a small-molecule inhibitor of AICAR transformylase homodimerization. Chembiochem. 2012;13(11):1628–34.
Zhu Y, Lu H, Zhang D, Li M, Sun X, Wan L, et al. Integrated analyses of multi-omics reveal global patterns of methylation and hydroxymethylation and screen the tumor suppressive roles of HADHB in colorectal cancer. Clin Epigenetics. 2018;10:30.
Briffa R, Um I, Faratian D, Zhou Y, Turnbull AK, Langdon SP, et al. Multi-scale genomic, transcriptomic and proteomic analysis of colorectal Cancer cell lines to identify novel biomarkers. PLoS One. 2015;10(12):e0144708.
Alexiou D, Karayiannakis AJ, Syrigos KN, Zbar A, Kremmyda A, Bramis I, et al. Serum levels of E-selectin, ICAM-1 and VCAM-1 in colorectal cancer patients: correlations with clinicopathological features, patient survival and tumour surgery. Eur J Cancer. 2001;37(18):2392–7.
Ngan CY, Yamamoto H, Seshimo I, Ezumi K, Terayama M, Hemmi H, et al. A multivariate analysis of adhesion molecules expression in assessment of colorectal cancer. J Surg Oncol. 2007;95(8):652–62.
Zlobec I, Lugli A, Baker K, Roth S, Minoo P, Hayashi S, et al. Role of APAF-1, E-cadherin and peritumoral lymphocytic infiltration in tumour budding in colorectal cancer. J Pathol. 2007;212(3):260–8.
Ito Y, Hart JR, Ueno L, Vogt PK. Abstract 3339: the regulatory subunit of PI3K, p85β, induces cellular transformation, enhanced cell proliferation and increased PI3K signaling. Cancer Res. 2014;74(19 Supplement):3339.
Danielsen SA, Eide PW, Nesbakken A, Guren T, Leithe E, Lothe RA. Portrait of the PI3K/AKT pathway in colorectal cancer. Biochim Biophys Acta. 2015;1855(1):104–21.
Acknowledgements
Not applicable.
Funding
None.
Author information
Authors and Affiliations
Contributions
GW contributed to the drafting of the article. BS contributed to the concept and design of the study. FW and ZM contributed to the interpretation of data, and the critical revision of the study methods. NW, CZ and JZ contributed to the critical revision of the article for relevant intellectual content. LZ and GW made critical revisions of the article for valuable intellectual content. All authors approved the final version of the article, including the authorship list.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All participating individuals provided informed consent. This study was approved by the ethics committee of the Fourth Hospital of Hebei Medical University. All the experiment protocol for involving humans was in accordance to guidelines of national/international/institutional or Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Clinical information of included datasets.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, G., Wang, F., Meng, Z. et al. Uncovering potential genes in colorectal cancer based on integrated and DNA methylation analysis in the gene expression omnibus database. BMC Cancer 22, 138 (2022). https://doi.org/10.1186/s12885-022-09185-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-022-09185-0