
Abstract
Colorectal adenomas form a biologically and clinically distinct intermediate stage in development of colorectal cancer (CRC) from normal colon epithelium. Only 5% of adenomas progress into adenocarcinomas, indicating that malignant transformation requires other biological alterations than those involved in adenoma formation. The present study aimed to explore which cancer-related biological processes are affected during colorectal adenoma-to-carcinoma progression and to identify key genes within these pathways that can serve as tumor markers for malignant transformation. The activity of 12 cancer-related biological processes was compared between 37 colorectal adenomas and 31 adenocarcinomas, using the pathway analysis tool Gene Set Enrichment Analysis. Expression of six gene sets was significantly increased in CRCs compared to adenomas, representing chromosomal instability, proliferation, differentiation, invasion, stroma activation, and angiogenesis. In addition, 18 key genes were identified for these processes based on their significantly increased expression levels. For AURKA and PDGFRB, increased mRNA expression levels were verified at the protein level by immunohistochemical analysis of a series of adenomas and CRCs. This study revealed cancer-related biological processes whose activities are increased during malignant transformation and identified key genes which may be used as tumor markers to improve molecular characterization of colorectal tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is the second leading cause of cancer mortality in the western world. CRC develops from normal colon epithelium through an adenoma precursor stage. Premalignant colon adenomas arise by disruption of the Wnt-signaling pathway, mostly by DNA mutations in, or (epigenetic) silencing of the APC tumor suppressor gene [1]. Additional mutations in genes like KRAS and P53, as well as specific chromosome copy number changes like 20q gain, further contribute to adenoma-to-carcinoma progression [2, 3]. It is estimated that only a small proportion of about 5% of adenomas will advance to the clinically more relevant stage of CRC, implying that the biology of adenoma formation from normal colon epithelium differs significantly from that of malignant transformation of colorectal adenomas into adenocarcinomas.
Biological processes that are frequently altered during carcinogenesis affect the behavior of tumor cells and tissues. Tumor cells exhibit increased proliferation and lack of apoptosis, overcome senescence, and may gain invasive and metastatic potential [4]. Expanding tumors become hypoxic and induce angiogenesis, while sustained angiogenesis results in poor vasculature and contributes to activation of stroma by serum exposure (wound response) [4, 5]. Moreover, effective immune responses are circumvented or avoided, for instance by forcing tumor-associated macrophages to generate an immune suppressive microenvironment [6]. Accumulation of (epi)genetic aberrations leads to changes in gene expression and affect the activity of cancer-related pathways. Microarray mRNA expression studies allow to compare gene expression at a genome-wide level and to explore the transcriptional programs that are turned on or off in tumors during progression from normal through premalignant stages to cancer. However, extracting such complex information from large datasets derived from heterogeneous biological samples has proven to be difficult. Pathway analysis programs such as Gene Set Enrichment Analysis (GSEA) can be of help here [7]. GSEA makes use of the fact that changes in biological characteristics require coordinate variation in expression of groups of genes, i.e., gene sets, which regulate biological activity. While absolute changes in expression levels of the majority of individual genes are often modest to small and do not reach statistical significance, GSEA allows to estimate groupwise variation in expression of predefined gene sets, indicative for pathway activity levels.
The concept that cancer development is the result of defects in multiple biological processes, e.g., as described by Hanahan and Weinberg, is well accepted [4]. The present study aims to examine the changes in the activity of these cancer-related biological processes during malignant transformation of colorectal adenomas into adenocarcinomas. We here report careful composition of predefined gene sets that represent various aspects of carcinogenesis and application of the GSEA pathway analysis program to compare their activity levels in benign and malignant colorectal lesions. Moreover, individual genes that differed most significantly within the respective pathways were identified.
Materials and methods
Microarray mRNA expression profiling of colorectal tumor tissue samples
Genomewide mRNA expression data were obtained by microarray analysis of 37 colorectal adenomas and 31 adenocarcinomas, as described previously [8]. The microarray expression data can be accessed through the Gene Expression Omnibus under accession number GSE8067 (http://www.ncbi.nlm.nih.gov/geo/). Genes with missing values in more than 20% of tumor samples were excluded from further analysis. Remaining missing values were imputed using k-nearest neighbor averaging, as implemented in the R-package “impute” [9].
Selection of gene sets representing cancer-related processes
The pathway analysis tool GSEA makes use of predefined gene sets. We aimed to examine which carcinogenic processes could be relevant to adenoma-to-carcinoma progression and, therefore, restricted GSEA analysis to carefully selected cancer-related gene sets. Cancer-related processes explored comprised: proliferation, cell cycle, apoptosis, tumor differentiation, tumor cell invasion, metastasis, chromosomal instability, and microenvironmental changes such as hypoxia, angiogenesis, immune response, tumor-associated macrophages, and tumor stroma activation. On the one hand, Gene Ontology (GO) terms were used to obtain gene sets from the NCBI Entrez Gene database. The following keywords yielded GO-derived gene sets: proliferation, apoptosis, cell cycle, differentiation, hypoxia, angiogenesis, and immune response. On the other hand, gene sets were collected of which altered coordinated expression in cancer had been observed in an experimental setting, by searching the NCBI PubMed database. Cancer-related experiment-derived gene sets included: proliferation [10], tumor differentiation [11], invasion [12], metastasis [13], chromosomal instability [14], hypoxia [15], angiogenesis [16], tumor-associated macrophages [17], and stroma activation [18]. Together, seven GO-derived gene sets and nine experiment-derived gene sets were selected for pathway analysis by GSEA. The total number of genes within each gene set and the number of genes covered by the microarray expression data are listed in Table 1. A complete list of genes within the 16 gene sets is provided in Supplementary Table 1. Supplementary Table 2 gives detailed information about the origin of the gene sets.
GSEA pathway analysis of gene expression
Pathway analysis of gene expression was performed using GSEA (v2.0, http://www.broad.mit.edu/gsea/) [7]. GSEA performs a competitive analysis of predefined gene sets, which is suited for examination of relatively heterogeneous biological samples. In brief, GSEA first ranks all genes analyzed by expression arrays according to their differential expression between two categories of samples, in this case, colorectal adenomas and carcinomas. Next, for each predefined gene set analyzed, GSEA calculates a pathway enrichment score that indicates to what extent these gene sets are enriched for the highest—(or lowest)—ranking genes. Default settings were used, except for the maximum size of gene sets, which was set to 1,500 to include all predefined gene sets for analysis. Thresholds for significance of the enrichment score were determined by permutation analysis (1,000 permutations). False discovery rate (FDR) q values < 0.05 were considered significant.
Identification of key genes for pathway activity
Individual genes from cancer-related gene sets for which mRNA expression levels differed most between colorectal adenomas and CRCs, were considered key genes for pathway activity. For individual genes within cancer-related gene sets that contributed to the enrichment score, p values were calculated using the Student’s t test with unequal variance. P values < 1e-5 (uncorrected for multiple testing) were considered significant.
Immunohistochemistry analysis of AURKA and PDGFRB
Immunohistochemical staining of aurora kinase A (AURKA) and platelet-derived growth factor receptor beta (PDGFRB) was performed on 4-μm thick paraffin sections of a series of nine colorectal adenomas and 10 adenocarcinomas. Sections were deparaffinized in xylene and rehydrated through a series of graded alcohol to water. Endogenous peroxidase was blocked with hydrogen peroxide (0.3% H2O2 in methanol) for 30 min. Antigen retrieval was performed by autoclave heating in 10 mM citrate buffer (pH 6.0). Antibodies directed against AURKA (mouse monoclonal NCL-L-AK2, Novocastra Laboratories, Newcastle upon Tyne, UK) or PDGFRB (rabbit polyclonal 28E1, Cell Signaling Technology, Danvers, USA) were incubated overnight at 4°C (1:50 dilution). AURKA staining was visualized using the Dako EnVision Kit-K4006 (Dako Cytomation, Copenhagen, Denmark) and PDGFRB staining using the PowerVision+ Kit (ImmunoLogic, Duiven, NL) according to the manufacturer’s recommendations. Sections were counterstained with Mayer's hematoxylin. Incubation without primary antibody was used as negative control.
Results
GSEA pathway analysis of colorectal adenomas and CRCs
Six cancer-related gene sets showed increased expression in CRCs compared to adenomas (FDR < 0.05), i.e., chromosomal instability, proliferation, differentiation, angiogenesis, stroma activation, and invasion (Table 1). Of these, the change in activity of the chromosomal instability gene set was most significant (FDR = 0.004).
Identification of key genes for pathway activity in adenoma-to-carcinoma progression
While the far majority of individual genes within the six gene sets involved in adenoma-to-carcinoma progression will exhibit modest, nonsignificant variation in gene expression between colorectal adenomas and adenocarcinomas, specific genes within these gene sets that do show large variation in mRNA expression levels may function as key genes for the activity of cancer-related pathways involved in malignant transformation. To identify such genes from the six differentially expressed cancer-related gene sets, individual genes were ranked according to their difference in mRNA expression between 37 colorectal adenomas and 31 CRCs. P values < 1e-5 were considered significant at the genome-wide level. This procedure yielded key genes for five out of six biological processes: AURKA, C20orf24, and TPX2 (chromosomal instability); PLK1 and CCNF (proliferation); ADRM1 and NUDT1 (differentiation); SSSCA1, ID3, LUM, FYCO1, RFC3, LOXL2, SVIL, KIAA0367, and NUDT1 (stroma activation); and SPARC, DCN, and PDGFRB (invasion). For the angiogenesis gene set, none of the individual genes passed the threshold for genome-wide significance. Table 2 lists all genes within the cancer-related processes that exhibited p values <1e-4. The magnitude of differential expression between colorectal adenomas and adenocarcinomas for genes from each of these processes, i.e., AURKA, PLK1, ADRM1, SSSCA1, SPARC, and PDGFRB, is illustrated with boxplots (Fig. 1).

Comparison of mRNA expression levels for key genes of pathway activity in colorectal adenomas and CRCs. Boxplots showing mRNA expression levels of 37 colorectal adenomas and 31 colorectal adenocarcinomas, based on oligonucleotide microarray expression data. Expression of key genes was significantly higher in CRC compared to adenomas (p values < 1e-5). a AURKA; b PLK1; c ADRM1; d SSSCA1; e SPARC; and f PDGFRB
AURKA and PDGFRB protein expression in adenomas and CRCs
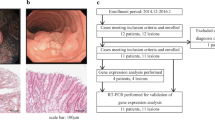
Most of the key genes for pathways involved in malignant transformation have previously been described to play a role in carcinogenesis, and some of them are considered potential cancer drug targets. For two such key genes for which good antibodies were available, AURKA (chromosomal instability gene set) and PDGFRB (invasion gene set), we aimed to validate their difference in mRNA expression between colorectal adenomas and CRCs at the protein level. Immunohistochemical stainings were performed on a series of nine colorectal adenomas and 10 adenocarcinomas. Four adenomas stained negative for AURKA, another four were weakly positive, while strong staining was observed in only one adenoma. In contrast, only two CRCs stained negative for AURKA, weak staining was observed for four CRCs, while strong AURKA expression was observed in the epithelial cells of four CRCs, consistent with previous observations [8]. For PDGFRB, staining was nearly absent in six out of nine adenomas, while three adenomas stained weakly positive. In contrast, PDGFRB protein expression was observed in the stromal compartment of nine out of 10 CRCs, four of which stained weakly positive and five of which stained strongly positive. Representative images for AURKA and PDGFRB staining are given in Fig. 2.

Comparison of protein expression levels for AURKA and PDGFRB in colorectal adenomas and CRCs. Immunohistochemical stainings for AURKA and PDGFRB confirmed overexpression of both proteins in CRCs compared to colorectal adenomas. AURKA staining can be found in the epithelial cells, while PDGFRB expression is observed in tumor stroma. Representative examples of AURKA and PDGFRB staining are shown for colorectal adenoma and carcinoma tissue. Digital images were obtained with a 20× objective (AURKA) and a 10× objective (PDGFRB)
Discussion
Colorectal adenoma formation and further progression into carcinomas is caused by accumulation of (epi)genetic alterations. As such, one might expect colorectal carcinogenesis to be a stochastic process in which, sooner or later, malignant progression is an inevitable event. However, biologically and clinically, colorectal adenomas form a distinct intermediate stage in CRC development from normal colon epithelium. It is estimated that only 5% of adenomas ever progress into adenocarcinomas, indicating that carcinoma formation from adenomas requires significantly different biological and, therefore, molecular alterations than those involved in adenoma formation from normal epithelium. Given the high prevalence of colorectal adenomas and the relatively low progression rate, one could argue that especially biological processes involved in adenoma-to-carcinoma progression are most relevant from a clinical point of view. We here report identification of six cancer-related biological processes whose activity is increased in CRCs compared to adenomas (Table 1) and a list of key genes whose increased mRNA expression levels are associated with malignant transformation (Table 2). For two of these genes, i.e., AURKA and PDGFRB, differential expression was verified at the protein level (Fig. 2).
The GSEA carcinogenic pathway analysis performed in the present study was restricted to a limited number of 16 carefully selected cancer-related gene sets for two reasons. First, although large databases that contain numerous predefined gene sets are available, such as the Molecular Signatures Database (www.broad.mit.edu/gsea/msigdb), none of these contain a well-defined subset of gene sets representing various biological aspects of carcinogenesis. Second, it is not recommended to perform GSEA using large groups of gene sets that are not relevant to the research question addressed, as this will increase the multiple testing problem and lead to unnecessary decrease of statistical power. Therefore, we set out to select gene sets representing cancer-related processes using two strategies, one based on Gene Ontology terms (seven gene sets) and one based on a PubMed literature search of in vitro and in vivo experimental data (nine gene sets). For four cancer-related processes (proliferation, differentiation, hypoxia, and angiogenesis) gene sets were obtained using both strategies, allowing to compare their value for GSEA carcinogenic pathway analysis. The experiment-derived “proliferation” and “differentiation” gene sets yielded a significant difference between adenomas and CRCs while their GO-derived equivalents did not. In contrast, the GO-derived “angiogenesis” gene set yielded a significant difference while the experiment-derived gene set did not. No significant differences were observed at all for the experiment- and GO-derived “hypoxia” gene sets. These data illustrate that both strategies revealed useful gene sets for GSEA carcinogenic pathway analysis. However, they also imply that optimal gene sets may not be available yet for all (colorectal) cancer-related processes.
The GSEA carcinogenic pathway analysis results indicated significantly different rates of chromosomal instability, proliferation, differentiation, angiogenesis, stroma activation, and invasion between colorectal adenomas and CRCs (Table 1). These results fit current knowledge about malignant transformation. Chromosomal instability increases the rate of genomic alterations, necessary to bypass the rate-limiting steps in carcinogenesis [19]. Analysis of chromosome copy number changes by comparative genomic hybridization has demonstrated that CRCs exhibit much more chromosomal instability than adenomas [3, 8]. The present data show that the chromosomal instability gene set changes highly significantly in activity during adenoma-to-carcinoma progression. This further emphasizes the importance of chromosomal instability in colorectal adenoma-to-carcinoma progression. Chromosomal instability might even be the driving force in tumor progression by initiating the changes in other cancer-related biological processes. Although the balance between proliferation and differentiation is already abnormal in adenomas, proliferation rates further increase during the adenoma–carcinoma sequence [20]. Angiogenesis is induced by growing tumors in an attempt to meet their increasing demand for oxygen and nutrients. Microvessel density, a widely used surrogate marker for angiogenesis, has been shown to be increased in CRCs compared to colorectal adenomas [21]. In comparison to adenomas, CRCs also contain much more tumor stroma, which is often composed of reactive tissue that resembles wounds that do not heal [5]. Interestingly, the amount of stroma differs widely among CRCs [22], and a high stroma percentage has been correlated with poor prognosis in CRC patients [23]. Hence, increased expression of the invasion gene set by CRCs fits the concept of adenoma-to-carcinoma progression.
For several cancer-related processes, no significant differences were revealed between adenomas and CRCs, i.e., for gene sets representing apoptosis, cell cycle, hypoxia, immune response, tumor-associated macrophages, and metastasis. One interpretation is that these biological processes are more relevant during formation of colorectal adenomas from normal colon epithelium, than during adenoma-to-carcinoma progression. Alternatively, although these biological processes could play a role in malignant transformation, the selected gene sets may not adequately represent the in vivo situation analyzed here. For instance, GO-derived gene sets are composed of groups of genes known to be involved in similar biological processes, irrespective of whether they actually function in a coordinated manner or not. In contrast, experiment-derived gene sets are composed of groups of genes that are coordinately expressed during certain biological processes; however, assumptions have been made about conservation of these gene sets across species, across tumor types and about the validity of extrapolation from in vitro to in vivo settings. Nevertheless, although our approach may underestimate the effects of some cancer-related processes in adenoma-to-carcinoma progression, the positively identified gene sets yield valuable information for further investigation, such as the identification of key genes for malignant transformation. Expression of individual genes within gene sets that were positively identified by GSEA yielded a list of these key genes for various carcinogenic processes that may be used for molecular characterization of series of tumor samples (Table 2). Some of these genes have been described to contribute to (colorectal) carcinogenesis. From the “chromosomal instability” gene set, AURKA and TPX2 (targeting protein for XKLP2) have been reported to interact with each other and to play a role in centrosome maturation and spindle formation [24]. Aberrant expression of TPX2 has been reported in breast, endometrial, and lung cancer and in neuroblastoma [25]. Furthermore, TPX2 overexpression at the protein level was found to be associated with poor prognosis in lung cancer [25]. AURKA, when overexpressed, induces centrosome amplification, aneuploidy, and cellular transformation in vitro [26]. In nasopharyngeal carcinoma, AURKA overexpression was correlated with clinical stage and invasiveness, and inhibition with small molecules or RNA interference reduced cell invasion in vitro [27]. From the “proliferation” gene set, polo-like kinase 1 (PLK1) is thought to play a role in spindle formation and in cell cycle progression during the G2 and M phase [28]. Interference with PLK1 expression decreases proliferation, induces apoptosis, and affects spindle assembly in vitro [29]. Moreover, down-modulation of PLK1 expression was found to inhibit growth of bladder cancer in mice [29]. Expression of PLK1 and CCNF (cyclin F), which contributes to the G2 to M phase transition, have been related to response to radio and chemotherapy [30, 31]. From the “invasion” gene set, secreted protein acidic and rich in cysteine (SPARC) (also known as osteonectin) is overexpressed in CRCs and induces proinvasive activity [32]. PDGFRB is upregulated within CRC tumor stroma, and blocking of PDGFRB signaling has been shown to inhibit colon tumor growth and metastasis [33].
Immunohistochemical analysis of a series of colorectal adenomas and adenocarcinomas was used to verify the expression of some of the key genes at the protein level, i.e., AURKA and PDGFRB (Fig. 2). CRCs exhibited more frequently more intense staining for both proteins than adenoma tissue. Therefore, both AURKA and PDGFRB may have the potential to be used as markers indicative for the activity level of “chromosomal instability” and “invasion,” respectively. For AURKA, protein staining was restricted to epithelial cells, indicating that AURKA influences CRC progression by its effect on tumor cells. PDGFRB staining was predominantly observed within tumor stroma, suggesting a stromal effect of PDGFRB on cancer progression. In this way, information on protein expression helps to put mRNA expression data into biological context.
In summary, GSEA was applied as a tool for pathway analysis of gene expression using a restricted number of gene sets representing cancer-related biological processes. Expression of six gene sets was increased in CRCs compared to adenomas, of which the chromosomal instability pathway was most prominent. Subsequently, key genes within these gene sets that exhibited significant differential expression were identified. Further research is required to explore whether these genes can be used as tumor markers for malignant transformation, and/or as drug targets for distinct carcinogenic pathways that contribute to colorectal adenoma-to-carcinoma progression.
References
Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7.
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67.
Hermsen M, Postma C, Baak J, Weiss M, Rapallo A, Sciutto A, et al. Colorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instability. Gastroenterology. 2002;123:1109–19.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9.
Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50.
Carvalho B, Postma C, Mongera S, Hopmans E, Diskin S, van de Wiel MA, et al. Terhaar Sive Droste JS, Craanen M, Schrock E, Ylstra B, Meijer GA: Multiple putative oncogenes at the chromosome 20q amplicon contribute to colorectal adenoma to carcinoma progression. Gut. 2009;58:79–89.
Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, et al. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17:520–5.
Whitfield ML, George LK, Grant GD, Perou CM. Common markers of proliferation. Nat Rev Cancer. 2006;6:99–106.
Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A. 2004;101:9309–14.
Jechlinger M, Grunert S, Tamir IH, Janda E, Ludemann S, Waerner T, et al. Expression profiling of epithelial plasticity in tumor progression. Oncogene. 2003;22:7155–69.
Li DQ, Hou YF, Wu J, Chen Y, Lu JS, Di GH, et al. Gene expression profile analysis of an isogenic tumour metastasis model reveals a functional role for oncogene AF1Q in breast cancer metastasis. Eur J Cancer. 2006;42:3274–86.
Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043–8.
Greijer AE, van der Groep P, Kemming D, Shvarts A, Semenza GL, Meijer GA, et al. Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1). J Pathol. 2005;206:291–304.
Hu J, Bianchi F, Ferguson M, Cesario A, Margaritora S, Granone P, et al. Gene expression signature for angiogenic and nonangiogenic non-small-cell lung cancer. Oncogene. 2005;24:1212–9.
Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood. 2006;107:2112–22.
Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, et al. van de RM, Botstein D, Brown PO: Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7.
Loeb LA. A mutator phenotype in cancer. Cancer Res. 2001;61:3230–9.
Lipkin M. Phase 1 and phase 2 proliferative lesions of colonic epithelial cells in diseases leading to colonic cancer. Cancer. 1974;34:suppl:878–88.
Kondo Y, Arii S, Furutani M, Isigami S, Mori A, Onodera H, et al. Implication of vascular endothelial growth factor and p53 status for angiogenesis in noninvasive colorectal carcinoma. Cancer. 2000;88:1820–7.
Fijneman RJ, Carvalho B, Postma C, Mongera S, van Hinsbergh VW, Meijer GA. Loss of 1p36, gain of 8q24, and loss of 9q34 are associated with stroma percentage of colorectal cancer. Cancer Lett. 2007;258:223–9.
Mesker WE, Junggeburt JM, Szuhai K, de Heer P, Morreau H, Tanke HJ, et al. The carcinoma-stromal ratio of colon carcinoma is an independent factor for survival compared to lymph node status and tumor stage. Cell Oncol. 2007;29:387–98.
De Luca M, Lavia P, Guarguaglini G. A functional interplay between Aurora-A, Plk1 and TPX2 at spindle poles: Plk1 controls centrosomal localization of Aurora-A and TPX2 spindle association. Cell Cycle. 2006;5:296–303.
Ma Y, Lin D, Sun W, Xiao T, Yuan J, Han N, et al. Expression of targeting protein for xklp2 associated with both malignant transformation of respiratory epithelium and progression of squamous cell lung cancer. Clin Cancer Res. 2006;12:1121–7.
Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–93.
Wan XB, Long ZJ, Yan M, Xu J, Xia LP, Liu L, et al. Inhibition of Aurora-A suppresses epithelial-mesenchymal transition and invasion by downregulating MAPK in nasopharyngeal carcinoma cells. Carcinogenesis. 2008;29:1930–7.
Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat Cell Biol. 2000;2:672–6.
Nogawa M, Yuasa T, Kimura S, Tanaka M, Kuroda J, Sato K, et al. Intravesical administration of small interfering RNA targeting PLK-1 successfully prevents the growth of bladder cancer. J Clin Invest. 2005;115:978–85.
Lyng H, Landsverk KS, Kristiansen E, DeAngelis PM, Ree AH, Myklebost O, et al. Response of malignant B lymphocytes to ionizing radiation: gene expression and genotype. Int J Cancer. 2005;115:935–42.
De Angelis PM, Fjell B, Kravik KL, Haug T, Tunheim SH, Reichelt W, et al. Molecular characterizations of derivatives of HCT116 colorectal cancer cells that are resistant to the chemotherapeutic agent 5-fluorouracil. Int J Oncol. 2004;24:1279–88.
Nguyen QD, De Wever O, Bruyneel E, Hendrix A, Xie WZ, Lombet A, et al. Commutators of PAR-1 signaling in cancer cell invasion reveal an essential role of the Rho-Rho kinase axis and tumor microenvironment. Oncogene. 2005;24:8240–51.
Kitadai Y, Sasaki T, Kuwai T, Nakamura T, Bucana CD, Fidler IJ. Targeting the expression of platelet-derived growth factor receptor by reactive stroma inhibits growth and metastasis of human colon carcinoma. Am J Pathol. 2006;169:2054–65.
Acknowledgements
This study was financially supported by the Cancer Center Amsterdam (AHS-H) and by an Aegon International Scholarship in Oncology (RJAF).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sillars-Hardebol, A.H., Carvalho, B., de Wit, M. et al. Identification of key genes for carcinogenic pathways associated with colorectal adenoma-to-carcinoma progression. Tumor Biol. 31, 89–96 (2010). https://doi.org/10.1007/s13277-009-0012-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-009-0012-1