
Abstract
Background
Triple-negative breast cancer (TNBC) is a highly heterogeneous subtype of breast cancer, showing aggressive clinical behaviors and poor outcomes. It urgently needs new therapeutic strategies to improve the prognosis of TNBC. Bioinformatics analyses have been widely used to identify potential biomarkers for facilitating TNBC diagnosis and management.
Methods
We identified potential biomarkers and analyzed their diagnostic and prognostic values using bioinformatics approaches. Including differential expression gene (DEG) analysis, Receiver Operating Characteristic (ROC) curve analysis, functional enrichment analysis, Protein-Protein Interaction (PPI) network construction, survival analysis, multivariate Cox regression analysis, and Non-negative Matrix Factorization (NMF).
Results
A total of 105 DEGs were identified between TNBC and other breast cancer subtypes, which were regarded as heterogeneous-related genes. Subsequently, the KEGG enrichment analysis showed that these genes were significantly enriched in ‘cell cycle’ and ‘oocyte meiosis’ related pathways. Four (FAM83B, KITLG, CFD and RBM24) of 105 genes were identified as prognostic signatures in the disease-free interval (DFI) of TNBC patients, as for progression-free interval (PFI), five genes (FAM83B, EXO1, S100B, TYMS and CFD) were obtained. Time-dependent ROC analysis indicated that the multivariate Cox regression models, which were constructed based on these genes, had great predictive performances. Finally, the survival analysis of TNBC subtypes (mesenchymal stem-like [MSL] and mesenchymal [MES]) suggested that FAM83B significantly affected the prognosis of patients.
Conclusions
The multivariate Cox regression models constructed from four heterogeneous-related genes (FAM83B, KITLG, RBM24 and S100B) showed great prediction performance for TNBC patients’ prognostic. Moreover, FAM83B was an important prognostic feature in several TNBC subtypes (MSL and MES). Our findings provided new biomarkers to facilitate the targeted therapies of TNBC and TNBC subtypes.
Similar content being viewed by others

Background
Judging from the immunohistochemistry (IHC) expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor2 (HER2), breast cancer can be broadly classified into luminal-like (luminal A/B-like), HER2-positive, and triple-negative [1,2,3,4]. Triple-negative (ER-, PR-, and HER2-) breast cancer (TNBC), a heterogeneous disease, comprises approximately 10–20% of all breast cancers [5,6,7,8] and can be further grouped into distinct subtypes by gene expression profiling [9]. TNBC has higher histopathological grade, proliferation index and risk of invasion than other breast cancer subtypes [10, 11], posing a great challenge for public health.
Due to the peculiar phenotype of TNBC, patients with the conventional anti-cancer treatment strategies may not show effective outcomes. Chemotherapy currently is the standard treatment [12,13,14], while TNBC patients frequently develop resistance to the chemotherapy drugs [15,16,17]. Over 50% are likely to recur after chemotherapy regimens [18, 19], which gives rise to great difficulty for the treatment of TNBC. Furthermore, the accurate diagnosis of early-stage TNBC is still tricky, and the advanced stage may accompany a poor prognosis [7, 10, 20]. Previous studies reported that the 5-year survival rate of TNBC patients dramatically decreased from stage II to stage III (from 76% to 45%) [21], which suggested that early diagnosis could improve the survival rate of TNBC [22]. Hence, it is urgent to find the specific and sensitive biomarkers to diagnose TNBC.
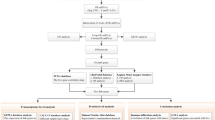
Recently, bioinformatics methods were widely used in cancer research and applied to study the molecular mechanisms of diseases [23,24,25,26,27,28]. The pathogenesis of TNBC may be a series of modifications of genetic, genomic and expression profiles [29]. Therefore, transcriptome based genetic perspective research is helpful to understand the occurrence and development of TNBC [30]. Biomarkers may have a great accurate prediction of metastatic behaviors in TNBC [31]. Furthermore, they can evaluate the diagnosis and prognosis of TNBC subtypes [32]. Overall, bioinformatics would contribute to the precise treatment of TNBC. In this study, transcriptome data were used to explore potential biomarkers of TNBC and TNBC subtypes. The workflow showed in Fig. 1.

The workflow for identification of potential diagnostic and prognostic biomarkers in triple-negative breast cancer
Methods
Acquisition of gene expression data and patient's clinical information
Gene expression microarray datasets were obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) according to the following requirements: (1) dataset was mRNA expression profile data; (2) the information of the platform annotation was accessible; (3) the expression data of microarray had been published. Therefore, the datasets including GSE115275 [33] and GSE62931 [34] were downloaded by GEO query package in R software (version 3.6.1, https://www.r-project.org). Meanwhile, mRNA sequencing data of 1247 samples of breast cancer and normal breast tissue were acquired from the TCGA database (http://portal.gdc.cancer.gov/). Subsequently, the data from TCGA and GEO datasets were filtered, and a total of 120 normal tissue samples, 176 TNBC samples, and 916 non-TNBC samples were finally brought into our study (Additional file 1: Table S1).
Differential expression gene analysis
Before differential expression gene (DEG) analysis, tumor purity estimation and principal component analysis (PCA) were used to evaluate the samples. The limma package was used to perform DEG analysis, and DEGs (FDR < 0.05 & |log2FC| ≥ 1) were considered to be significantly changed. Then, the up- and down- regulated DEGs were further verified in sequencing data of TCGA.
Receiver operating characteristic curve analysis and enrichment analysis
The plotROC package was employed to predict the ability of DEGs to identify TNBC from breast cancer via Receiver Operating Characteristic (ROC) curve analysis. DEGs with Area Under the Curve (AUC) higher than 0.7 were further used for GO and KEGG (www.kegg.jp/kegg/kegg1.html) [35, 36] pathway enrichment analyses through FunRich (version 3.1.3, http://www.funrich.org) software [37].
Protein-protein interaction network and module analysis
In the meanwhile, DEGs with discriminative ability to TNBC were used to download protein interaction information from STRING (https://string-db.org/). After that, Cytoscape (version 3.6.0, https://cytoscape.org) software was employed to build the protein-protein interaction (PPI) network. Molecular Complex Detection (MCODE) plug-in was used to detect modules in the network [38, 39]. Models with score ≥ 5 were considered for further enrichment analysis, which could help to explore the potential biological functions of DEGs. Subsequently, the CytoNCA was applied to evaluate betweenness centrality (BC), degree centrality (DC), and closeness centrality (CC) of the nodes in the network modules [40].
Univariate and multivariate cox regression analysis
Survival package in R software was performed to univariate Cox regression analysis of DEGs, and DEGs with p < 0.05 were considered as prognosis signatures. Then, multivariate Cox regression models for the DFI (DFI defined as the time between the treatment of primary cancer and the development of regional metastases [41]) and PFI (PFI defined as the time from randomization to the first documentation of objective tumor progression or death [42]) were established based on these DEGs. And the risk score was calculated according to the following formula: risk score = \( {\sum}_{i=1}^n\left({coef}_i\ast {Expr}_i\right) \), where coef was the regression coefficient of genes and expr represented the gene expression values. According to the median risk score, TNBC samples were divided into high- and low-risk groups. Besides, the time-dependent ROC curve was plotted, which presented the predictive accuracy of multivariate Cox regression models. Furthermore, rms package was used to integrate the clinical information (including age, pathological stage, menopause and ethnicity) into the multivariate Cox regression model, and the sensitivity and specificity of the model were calculated.
Survival analyses of TNBC subtypes
TNBCtype (https://cbc.app.vumc.org/tnbc/) tool [43] and the TNBC subtype-specific genes [44] were respectively used to identify TNBC subtypes. Six distinct subtypes were discovered by TNBCtype, including two basal-like (BL1 and BL2), an immunomodulatory (IM), a mesenchymal (M), a mesenchymal stem-like (MSL), and a luminal androgen receptor (LAR). And four TNBC clusters, such as Luminal Androgen Receptor (LAR), mesenchymal (MES), basal-like immunosuppressed (BLIS), and basal-like immune-activated (BLIA) were obtained based on TNBC subtype-specific genes. Finally, the prognostic value of TNBC prognostic factors was evaluated according to these classifications.
Results
Identification of DEGs in TNBC tissue samples
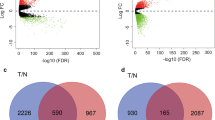
The tumor purity of 1092 breast cancer tissues were between 0.159 and 0.994, and 56.4% of them had a purity value greater than the mean of 0.741 (Fig. 2a-b). Additionally, the results of PCA demonstrated that in the selected datasets, the experimental group and the control group could be well distinguished (Fig. 2c-f), which suggested the selected samples were suitable for further analysis. As shown in Fig. 2g-j, 2158 DEGs were identified between TNBC and normal breast tissues (FDR < 0.05 and |log2FC| ≥ 1) were identified in GSE115275, including 1291 up-regulated genes and 867 down-regulated genes, while 1170 DEGs between TNBC and non-TNBC (558 up-regulated genes and 612 down-regulated genes) were obtained from GSE62931. After validation with the TCGA database, 991 DEGs (482 up-regulated genes and 509 down-regulated genes) between TNBC and non-TNBC, and 997 DEGs (491 up-regulated genes and 506 down-regulated genes) between TNBC and non-TNBCnormal breast tissues were screened out. The results were shown in Fig. 2k-n, which illustrated that these DEGs could be used to cluster different groups of samples. Finally, 125 common genes were obtained by intersecting the DEGs between TNBC and normal breast tissues and DEGs between TNBC and non-TNBC (Additional file 2: Table S2).

The results of DEGs analyses in GSE62931, GSE115275 and TCGA cohort. a Boxplot of tumor purity in TCGA, GES115275 and GES62931. b Histograms of tissues’ tumor purity. PCA plot of c GSE115275, d normal and TNBC samples from TCGA, e GSE62931, f non-TNBC and TNBC samples from TCGA. Volcano plot of g GSE115275, h normal and TNBC samples from TCGA, (i) GSE62931, (j) non-TNBC and TNBC samples from TCGA. Upregulated genes and downregulated genes are represented by red and blue dots in the volcano map, however, the genes having no obvious difference are represented by gray. Heatmap of (k) GSE115275, (l) normal and TNBC samples from TCGA, (m) GSE62931, (n) non-TNBC and TNBC samples from TCGA
ROC analysis of DEGs
For further screening, the verification efficiency of the 125 DEGs was measured using the ROC curve analysis. Among them, 121 DEGs had the good talent to distinguish normal breast tissues and TNBC samples, while 109 DEGs had superior performance in distinguishing between non-TNBC samples and TNBC samples. The intersection of these DEGs was taken to get 105 heterogeneous-related genes that could specifically identify TNBC (Additional file 3: Table S3). As shown in Fig. 3a, based on the AUC values, the top 5 genes were AURKA, ADAM33, CDCA8, CDCA3 and NUF2. Moreover, the expression of these genes in non-TNBC and TNBC was higher than that in normal tissues (Fig. 3b).

The top ten heterogeneous-related genes screened out from DEGs
Enrichment analysis of heterogeneous-related genes
We then performed GO functional annotation and KEGG pathway enrichment analysis of the heterogeneous-related genes (Additional file 4: Table S4) were performed then. As shown in Fig. 4a, KEGG pathway enrichment analysis revealed that they were primarily enriched in ‘cell cycle, mitotic’, ‘mitotic M-M/G1 phases’, and ‘DNA replication’ related pathways. In terms of biological process, these genes were significantly enriched in ‘spindle assembly’, ‘chromosome segregation’ and ‘cell communication’ (Fig. 4b). As for cellular composition, the heterogeneous-related genes were significantly related to ‘chromosome, centromeric region’, ‘microtubule’, and ‘outer kinetochore of condensed chromosome’ (Fig. 4c). Additionally, these genes were mainly involved in ‘Pprotein serine/threonine kinase activity’, ‘motor activity’ and ‘lipoprotein receptor activity’ of molecular functions (Fig. 4d).

GO and KEGG analysis of heterogeneous-related genes. a Biological pathway of heterogeneous-related genes. b Biological process of heterogeneous-related genes. c Cellular component of heterogeneous-related genes. d Molecular function of heterogeneous-related genes. e PPI network of heterogeneous-related genes. Upregulated genes are represented by red nodes and downregulated genes are represented by blue nodes. f Module, MCODE score ≥ 5 1 of PPI network. g-h Pathway analysis of module 1. i-j Functional enrichment analysis of module 1
PPI network establishment and pathway analysis of network module
Through inputting the 105 heterogeneous-related genes into the STRING, the PPI network of 79 genes was constructed (combined score > 0.4, Fig. 4e). The highest-scoring module (module 1), a cluster with score = 50.755, was detected by MCODE, which includingincluded 54 genes. Furthermore, the hub nodes (DLGAP5, KIF20A, CENPA, NUF2, and CDCA8) of module 1 were determined by CytoNCA (Fig. 4f). Finally, according to enrichment analysis, the heterogeneous-related genes in module 1 were enriched in ‘cell cycle’ and ‘oocyte meiosis’ related pathways, as well as ‘chromosome segregation’ and ‘mitotic nuclear division’ related gene functions (Fig. 4g-j).
Univariate cox regression analysis and identification of prognostic factors
To identify prognostic features among the 105 heterogeneous-related genes in TNBC patients’ DFI and PFI, the univariate Cox regression models and plotted Kaplan-Meier curves were constructed accordingly. As shown in Fig. 5a-b and Additional file 5: Table S5, four genes (FAM83B, KITLG, CFD and RBM24) were significantly (p < 0.05) correlated with the DFI of 109 TNBC patients and five genes (FAM83B, EXO1, S100B, TYMS and CFD) significantly (p < 0.05) affected the PFI of 120 TNBC patients. As shown in Additional file 6: Table S6, among four critical clinical parameters (age, menopause, ethnicity, and the pathological stage), and found that the pathological stage was a prognostic factor in both DFI and PFI models (p < 0.05).

The result of Cox regression analysis of TNBC patients in the DFI and PFI models. a Kaplan-Meier curves of analysis of DFI for FAM83B, KITLG, RBM24 and CFD. b Kaplan-Meier curves of PFI for FAM83B, EXO1, S100B, TYMS and CFD. c Volcano plot of risk score of high and low risk TNBC patients in the DFI model. d Scatter plot of survival status and time of TNBC patients in the DFI model. e Expression of FAM83B, KITLG and RBM24 in high-risk and low-risk groups in the DFI model. f Kaplan-Meier curve for DFI in low-risk and high-risk groups. g 3-year, 5-year and 7-year time-dependent ROC curves of the DFI model. h Volcano plot of risk score of TNBC patients in the PFI model. i Scatter plot of survival status and time of TNBC patients in the PFI model. j Expression of FAM83B and S100B in high-risk and low-risk groups in the PFI model. k Kaplan-Meier curve for PFI in low-risk and high-risk groups. l Time-dependent ROC for 3-year, 5-year and 7-year PFI in TNBC patients 3-year, 5-year and 7-year time-dependent ROC curves of the PFI model
Multivariate cox regression analysis and establishment of prognostic risk model
Based on the results from the above analysis, multivariate Cox regression analysis was performed to establish a DFI model with the four prognostic features. The following formula was used to calculate the risk score: risk score = expression of FAM83B ∗ (− 1.584) + expression of KITLG ∗ 1.225 + expression of RBM24 ∗ 1.182. After ranking the risk scores, the result showed that the high-risk group showed high expression of KITLG and RBM24 as well as low expression of FAM83B (Fig. 5c-e). As shown in Fig. 5f, survival analysis manifested that the high-risk groups exhibited worse prognosis than the low-risk groups. Furthermore, the corresponding AUC value of the risk score for the 3-year, 5-year, and 7-year DFI were 0.711, 0.702, and 0.717, respectively (Fig. 5g).
Besides, we also performed multivariate Cox regression analysis was performed according to the prognostic factors of PFI. The calculation formula was as follows: risk score = expression of FAM83B ∗ (− 1.437) + expression of S100B ∗ (− 1.020). As shown in Fig. 5h-k, high risk scores were associated with by poor prognosis. Meanwhile, the AUC values of 3-, 5- and 7-year (0.758, 0.709 and 0.733) were shown in Fig. 5l, which suggested that the risk score had a good predictive ability for prognosis.
Clinic correlation and of prognostic factors
Nomograms were plotted to further evaluate the predictive significance of the clinical prognosis factor, combining the results of multivariate Cox regression analysis (Fig. 6a-b). As shown in Fig. 6c, the AUC values of the 3-, 5-year and 7-year DFI were 0.877, 0.836 and 0.829, respectively, and 0.775, 0.751 and 0.757 for PFI for 3-, 5- and 7-years, respectively (Fig. 6d). It indicated that the nomograms had a good predictive performance of TNBC patients.

Establishment and evaluation of nomogram model. a-b The nomograms for DFI and PFI. c-d 3-year, 5-year and 7-year time-dependent ROC curves of the nomogram model
Survival analyses of TNBC subtypes
Based on the Non-negative Matrix Factorization (NMF) algorithm, TNBC tissues were divided into four clusters stably (Fig. 7a-b). As shown in Fig. 7c, (1) all MSL and LAR TNBC subtypes were included in MSL and LAR clusters respectively; (2) most IM and M were included in BLIA and BLIS clusters respectively; (3) BL1 appeared in both BLIA and BLIS. The result of survival analysis showed that BLIA had a better prognosis than other TNBC subtypes, whereas MSL and MES clusters were associated with poor prognosis (Fig. 7d-g). Moreover, the expression differences of FAM83B, KITLG, RBM24 and S100B between different classifications were shown in Fig. 7h-i. Of note, compared with LAR (both in TNBC subtypes and TNBC clusters), S100B was significantly up-regulated in IM (BLIA), MSL (MES) and M (BLIS). Thus, S100B might be used for TNBC subtypes identification. Not only that, our results showed that FAM83B significantly affected the prognosis of both MSL and MSE (p < 0.05, Fig. 7j-m). Moreover, KITLG, RBM24 and S100B also had an effect on the prognosis of some TNBC subtypes with no significant difference (0.05 < p < 0.2, Additional file 7: Fig. S1).

Survival analysis of TNBC subtypes. a Cophenetic and dispersion metrics for 2–9 clusters. b Consensus plots for rank basis four NMF clusters. c Heatmap of gene expression of four clusters. d-e Kaplan-Meier curves for DFI and PFI in different TNBC types. f-g Kaplan-Meier curves for DFI and PFI in different clusters. h-i Expression of FAM83B, KITLG, RBM24 and KITLG in different TNBC types and clusters. *p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001. (j-m) DFI and PFI Kaplan-Meier curves of FAM83B
Discussion
TNBC displays a worse prognosis than other breast cancer subtypes on account of more aggressive behaviors, higher risk of recurrences and the absence of targeted therapeutic options [10, 11, 45, 46]. The present study, we focused on heterogeneous-related genes of TNBC to find new biomarkers, which might facilitate the development of TNBC treatments.
Our result showed that 105 heterogeneous-related genes had good performances in distinguishing TNBC from other breast cancer subtypes. The top 5 of them were AURKA, ADAM33, NUF2, CDCA3 and CDCA8. AURKA is a key regulator of mitosis and a kinase that promotes cancer migration and invasion [47]. It might facilitate TNBC cell proliferation by regulating the ‘cell cycle, mitotic’ signaling pathway (Fig. 4a) and played a major role in promoting TNBC metastatic phenotype [48]. It’s believed that ADAM33 may inhibit aggressiveness and metastasis inof TNBC, thus it has the function of tumor suppression [49, 50]. NUF2 may regulate the carcinogenesis and progression of breast cancer via ‘cell cycle’ related pathways [51]. Recent studies also showed that the expression level of NUF2 was higher in TNBC than that in non-TNBC [52]. Cell division cycle-associated (CDCA) genes could lead to malignancy through dislocating cell division [53,54,55]. High expression of CDCA3 and CDCA8 had been found in breast cancer, resulting in a poor prognosis [55]. Of note, compared with normal tissues, these five genes showed the same regulation trend in TNBC and non-TNBC, especially biggish in TNBC (Fig. 3b), which might lead to higher malignancy of TNBC andthan other phenotypes.
DFI and PFI have crucial roles in assessing cancer prognosis once the disease has recurred [56, 57]. Thus, DFI and PFI were considered as outcome indicators in this study. In the DFI model, up-regulated KITLG, RBM24 and CFD were correlated with poor prognoses, while FAM83B exhibited the contrary effect. Moreover, high expression of FAM83B, EXO1, S100B and TYMS prolonged the PFI, while CFD shortened the PFI of TNBC patients. A recent study suggested FAM83B could inhibit cancer chemotherapy resistance through inhibiting the Wnt pathway [58]. However, FAM83B might activate the MAPK and PI3K/Akt pathways [59, 60]. Up-regulated FAM83B could cause poor OS in pancreatic ductal adenocarcinoma (PDAC) [61] and non-small cell lung cancer (NSCLC) [62]. Overall, little is known about the role of FAM83B in cancers, especially in TNBC. Our result showed that FAM83B could be a prognostic feature of both TNBC and TNBC subtypes. Therefore, it is urgently required to explore the mechanism between FAM83B and TNBC development. There was a negative correlation between FAM83B and CFD (Additional file 8: Fig. S2), which indicated that they might play opposite roles in the progression of TNBC. Complement factor D (CFD) was a rate-limiting enzyme [63], implicating in the alternative complement pathway activation and involving in an innate immune pathway in numerous cancers [63,64,65]. Like CFD, KITLG could also be considered as a predictive marker for multiple cancers [66, 67]. Our findings showed CFD and KITLG (an immunostimulatory molecule [68]) enriched in the ‘immune system’ pathway (Additional file 4: Table S4). This pathway could influence breast tumor progression and immune surveillance [69]. Thus, it could be seen that they might synergistically promote breast cancer progression. S100B is an inflammatory mediator [70]. The previous study had demonstrated that a high S100B expression predicted a good distant metastases-free survival in all patients with breast cancer [70], which was in line with our result.
EXO1 and TYMS were closely correlated with CFD (Additional file 8: Fig. S2). TYMS plays a crucial role in DNA synthesis and repair [71]. And a recent study demonstrated that a high TYMS level predicted a good prognosis in TNBC patients [72]. EXO1 is a core gene of DNA metabolism [73]. Its polymorphism is related to clinical outcome and susceptibility of multiple tumors [74, 75]. Qi, L. et al. [76] suggested that a higher expression level of EXO1 came with a shorter OS in TNBC. We also confirmed the relationship between EXO1 expression and OS in TNBC (Additional file 9: Table S7), and the result was the same as that of Qi, L. et al. It has been reported that PFI might not be positive correlated with OS [77], which seems to be reasonable that the high level of EXO1 accompanied by long PFI in our result. Collectively, TYMS might be considered as a potential biomarker of TNBC, but the role of EXO1 was waiting for the further reveal.
RBM24 might relate to ‘regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolism’ of biological process (Additional file 4: Table S4). What’s more, several studies demonstrated that RBM24 might regulate the stability of mRNA transcripts in different human cancer cell lines [78, 79]. Thus, it could be speculated that RBM24 might have an effect on promoting cancer. However, the association between RBM24 and TNBC is not yet well understood. It warranted further investigation regarding its precise molecular mechanism.
In conclusion, our study provided potential biomarkers for TNBC and TNBC subtypes, which might provide new treatment strategies for the research and clinical treatment of TNBC. However, this study had several limitations as well. First of all, we were unable to determine the reasons for the significant changes of the heterogeneous-related genes in TNBC patients (i.e., we cannot distinguish between expression changes that are due to tumor-specific mutations or epigenetic modification). Secondly, because the publicly available gene microarray from different databases could inevitably introduce bias, our conclusions need to be verified in more datasets. Finally, the findings of the present study need to demonstrate in vivo and in vitro experiments.
Conclusions
In conclusion, we identified nine (four for DFI, five for PFI) of 105 heterogeneous-related genes were identified that acted as promising diagnostic and prognostic biomarkers in patients with TNBC via integrated bioinformatics analysis. It is worth mentioning that FAM83B might be further used to predict the prognosis of MSL and MES subtypes. Both DFI and PFI models presented great performances in predicting 3-, 5-, and 7-year prognoses. Our findings may provide new diagnostic and prognostic biomarkers to facilitate the targeted therapies of TNBC and TNBC subtypes.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the Gene Expression Omnibus (GEO) https://www.ncbi.nlm.nih.gov/geo/), R software (version 3.6.1, https://www.r-project.org), The Cancer Genome Atlas (TCGA) http://portal.gdc.cancer.gov/), The Gene Ontology (GO) http://geneontology.org/), KEGG database (https://www.kegg.jp/kegg/kegg1.html), FunRich (version 3.1.3, http://www.funrich.org), STRING (https://string-db.org/), Cytoscape (version 3.6.0, https://cytoscape.org), and TNBCtype (https://cbc.app.vumc.org/tnbc/).
The RNA-seq raw data are available at the GEO database under accession number GSE115275 and GSE62931.
Abbreviations
- TNBC:
-
Triple-negative breast cancer
- DEG:
-
Differential expression gene
- ROC:
-
Receiver operating characteristic
- PPI:
-
Protein-protein interaction
- DFI:
-
Disease-free interval
- PFI:
-
Progression-free interval
- IHC:
-
immunohistochemistry
- PR:
-
Progesterone receptor
- ER:
-
Estrogen receptor
- HER2:
-
Human epidermal growth factor receptor2
- GEO:
-
Gene Expression Omnibus
- TCGA:
-
The Cancer Genome Atlas
- PCA:
-
Principal component analysis
- AUC:
-
Area under the curve
- MCODE:
-
Molecular Complex Detection
- DC:
-
Degree centrality
- CC:
-
Closeness centrality
- BC:
-
Betweenness centrality
- BL1/2:
-
Basal-like 1/2
- IM:
-
Immunomodulatory
- M:
-
Mesenchymal
- MSL:
-
Mesenchymal stem–like
- LAR:
-
Luminal androgen receptor
- BLIS:
-
Basal-like immunosuppressed
- BLIA:
-
Basal-like immune-activated
- NMF:
-
Non-negative Matrix Factorization
- CDCA:
-
Cell division cycle-associated
- PDAC:
-
Pancreatic ductal adenocarcinoma
- NSCLC:
-
Non-small cell lung cancer
- CFD:
-
Complement factor D
References
de Ronde JJ, Hannemann J, Halfwerk H, Mulder L, Straver ME, Vrancken Peeters MJ, et al. Concordance of clinical and molecular breast cancer subtyping in the context of preoperative chemotherapy response. Breast Cancer Res Treat. 2010;119(1):119–26. https://doi.org/10.1007/s10549-009-0499-6.
Lee A, Moon BI, Kim TH. BRCA1/BRCA2 pathogenic variant breast Cancer: treatment and prevention strategies. Ann Lab Med. 2020;40(2):114–21. https://doi.org/10.3343/alm.2020.40.2.114.
Fan M, Chen J, Gao J, Xue W, Wang Y, Li W, et al. Triggering a switch from basal- to luminal-like breast cancer subtype by the small-molecule diptoindonesin G via induction of GABARAPL1. Cell Death Dis. 2020;11(8):635. https://doi.org/10.1038/s41419-020-02878-z.
Vagia E, Mahalingam D, Cristofanilli M: The Landscape of Targeted Therapies in TNBC. Cancers 2020;12(4):916.
Chuan T, Li T, Yi C. Identification of CXCR4 and CXCL10 as potential predictive biomarkers in triple negative breast Cancer (TNBC). Med Sci Monitor. 2020;26:e918281.
Rigiracciolo DC, Nohata N, Lappano R, Cirillo F, Talia M, Scordamaglia D, Gutkind JS, Maggiolini M: IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells. Cells 2020;9(4):1010.
De-la-Cruz-Ku G, Luyo M, Morante Z, Enriquez D, Möller MG, Chambergo-Michilot D, et al. Triple-negative breast cancer in Peru: 2000 patients and 15 years of experience. PLoS One. 2020;15(8):e0237811. https://doi.org/10.1371/journal.pone.0237811.
Morris GJ, Naidu S, Topham AK, Guiles F, Xu Y, McCue P, et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute's surveillance, epidemiology, and end results database. Cancer. 2007;110(4):876–84. https://doi.org/10.1002/cncr.22836.
Lehmann BD, Jovanović B, Chen X, Estrada MV, Johnson KN, Shyr Y, et al. Refinement of triple-negative breast Cancer molecular subtypes: implications for Neoadjuvant chemotherapy selection. PLoS One. 2016;11(6):e0157368. https://doi.org/10.1371/journal.pone.0157368.
Li HY, Liang JL, Kuo YL, Lee HH, Calkins MJ, Chang HT, et al. miR-105/93-3p promotes chemoresistance and circulating miR-105/93-3p acts as a diagnostic biomarker for triple negative breast cancer. Breast Cancer Res. 2017;19(1):133. https://doi.org/10.1186/s13058-017-0918-2.
Ryan D, Paul BT, Koziol J, ElShamy WM. The pro- and anti-tumor roles of mesenchymal stem cells toward BRCA1-IRIS-overexpressing TNBC cells. Breast Cancer Res. 2019;21(1):53. https://doi.org/10.1186/s13058-019-1131-2.
Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–81. https://doi.org/10.1200/JCO.2007.14.4147.
Tan DS, Marchió C, Jones RL, Savage K, Smith IE, Dowsett M, Reis-Filho JS: Triple negative breast cancer: molecular profiling and prognostic impact in adjuvant anthracycline-treated patients. Breast Cancer Res Treat 2008, 111(1):27–44, 1, DOI: https://doi.org/10.1007/s10549-007-9756-8.
Rastelli F, Biancanelli S, Falzetta A, Martignetti A, Casi C, Bascioni R, et al. Triple-negative breast cancer: current state of the art. Tumori. 2010;96(6):875–88. https://doi.org/10.1177/548.6505.
Chen J, Zhu M, Zou L, Xia J, Huang J, Deng Q, et al. Long non-coding RNA LINC-PINT attenuates paclitaxel resistance in triple-negative breast cancer cells via targeting the RNA-binding protein NONO. Acta Biochim Biophys Sin. 2020;52(8):801–9. https://doi.org/10.1093/abbs/gmaa072.
Mir M, Khan H, Mehraj U, Nisar S, Bhat B, Wani N. Targeting different pathways using novel combination therapy in triple negative breast Cancer. Curr Cancer Drug Targets. 2020;20(8):586–602. https://doi.org/10.2174/1570163817666200518081955.
Sharma P. Biology and Management of Patients with Triple-Negative Breast Cancer. Oncologist. 2016;21(9):1050–62. https://doi.org/10.1634/theoncologist.2016-0067.
Xiong G, Stewart RL, Chen J, Gao T, Scott TL, Samayoa LM, et al. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1α stabilization and TNBC chemoresistance. Nat Commun. 2018;9(1):4456. https://doi.org/10.1038/s41467-018-06893-9.
O'Reilly EA, Gubbins L, Sharma S, Tully R, Guang MH, Weiner-Gorzel K, et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015;3:257–75. https://doi.org/10.1016/j.bbacli.2015.03.003.
Sporikova Z, Koudelakova V, Trojanec R, Hajduch M. Genetic markers in triple-negative breast Cancer. Clin Breast Cancer. 2018;18(5):e841–50. https://doi.org/10.1016/j.clbc.2018.07.023.
Polyak K, Metzger Filho O. SnapShot: breast cancer. Cancer Cell. 2012;22(4):562–562.e561.
Soundararajan R, Fradette JJ, Konen JM, Moulder S, Zhang X, Gibbons DL, Varadarajan N, Wistuba, II, Tripathy D, Bernatchez C et al: Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019;11(5):714.
Li S, Yang R, Sun X, Miao S, Lu T, Wang Y, et al. Identification of SPP1 as a promising biomarker to predict clinical outcome of lung adenocarcinoma individuals. Gene. 2018;679:398–404. https://doi.org/10.1016/j.gene.2018.09.030.
Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW et al: The chromatin accessibility landscape of primary human cancers. Science (New York, NY) 2018;362(6413):eaav1898.
Xu M, Li Y, Li W, Zhao Q, Zhang Q, Le K, et al. Immune and Stroma related genes in breast Cancer: a comprehensive analysis of tumor microenvironment based on the Cancer genome atlas (TCGA) database. Front Med. 2020;7:64. https://doi.org/10.3389/fmed.2020.00064.
Li MX, Jin LT, Wang TJ, Feng YJ, Pan CP, Zhao DM, et al. Identification of potential core genes in triple negative breast cancer using bioinformatics analysis. OncoTargets Ther. 2018;11:4105–12. https://doi.org/10.2147/OTT.S166567.
Liu X, Wu J, Zhang D, Bing Z, Tian J, Ni M, et al. Identification of potential key genes associated with the pathogenesis and prognosis of gastric Cancer based on integrated bioinformatics analysis. Front Genet. 2018;9:265. https://doi.org/10.3389/fgene.2018.00265.
Zeng F, Fu J, Hu F, Tang Y, Fang X, Zeng F, et al. Identification of key pathways and genes in response to trastuzumab treatment in breast cancer using bioinformatics analysis. Oncotarget. 2018;9(63):32149–60. https://doi.org/10.18632/oncotarget.24605.
Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007;7(9):659–72. https://doi.org/10.1038/nrc2193.
Califf RM. Biomarker definitions and their applications. Exp Biol Med (Maywood, NJ). 2018;243(3):213–21.
Li G, Hu J, Hu G. Biomarker studies in early detection and prognosis of breast Cancer. Adv Exp Med Biol. 2017;1026:27–39. https://doi.org/10.1007/978-981-10-6020-5_2.
Pedersen MH, Hood BL, Ehmsen S, Beck HC, Conrads TP, Bak M, et al. CYPOR is a novel and independent prognostic biomarker of recurrence-free survival in triple-negative breast cancer patients. Int J Cancer. 2019;144(3):631–40. https://doi.org/10.1002/ijc.31798.
Tang L, Chen Y, Tang X, Wei D, Xu X, Yan F. Long noncoding RNA DCST1-AS1 promotes cell proliferation and metastasis in triple-negative breast Cancer by forming a positive regulatory loop with miR-873-5p and MYC. J Cancer. 2020;11(2):311–23. https://doi.org/10.7150/jca.33982.
Boac BM, Abbasi F, Ismail-Khan R, Xiong Y, Siddique A, Park H, et al. Expression of the BAD pathway is a marker of triple-negative status and poor outcome. Sci Rep. 2019;9(1):17496. https://doi.org/10.1038/s41598-019-53695-0.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51. https://doi.org/10.1002/pro.3715.
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49(D1):D545–d551. https://doi.org/10.1093/nar/gkaa970.
Pathan M, Keerthikumar S, Chisanga D, Alessandro R, Ang CS, Askenase P, et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J Extracellular Vesicles. 2017;6(1):1321455. https://doi.org/10.1080/20013078.2017.1321455.
Lin CY, Lee TL, Chiu YY, Lin YW, Lo YS, Lin CT, et al. Module organization and variance in protein-protein interaction networks. Sci Rep. 2015;5(1):9386. https://doi.org/10.1038/srep09386.
Otasek D, Morris JH, Bouças J, Pico AR, Demchak B. Cytoscape automation: empowering workflow-based network analysis. Genome Biol. 2019;20(1):185. https://doi.org/10.1186/s13059-019-1758-4.
Tang Y, Li M, Wang J, Pan Y, Wu FX. CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Bio Systems. 2015;127:67–72. https://doi.org/10.1016/j.biosystems.2014.11.005.
Höppener DJ, Nierop PMH, van Amerongen MJ, Olthof PB, Galjart B, van Gulik TM, et al. The disease-free interval between resection of primary colorectal malignancy and the detection of hepatic metastases predicts disease recurrence but not overall survival. Ann Surg Oncol. 2019;26(9):2812–20. https://doi.org/10.1245/s10434-019-07481-x.
Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med. 2015;372(7):621–30. https://doi.org/10.1056/NEJMoa1406470.
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. https://doi.org/10.1172/JCI45014.
Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21(7):1688–98. https://doi.org/10.1158/1078-0432.CCR-14-0432.
Prasad S, Efird JT, James SE, Walker PR, Zagar TM, Biswas T. Failure patterns and survival outcomes in triple negative breast cancer (TNBC): a 15 year comparison of 448 non-Hispanic black and white women. SpringerPlus. 2016;5(1):756. https://doi.org/10.1186/s40064-016-2444-6.
Battisti NML, Okonji D, Manickavasagar T, Mohammed K, Allen M, Ring A. Outcomes of systemic therapy for advanced triple-negative breast cancer: A single centre experience. Breast (Edinburgh, Scotland). 2018;40:60–6.
Wang X, Lu N, Niu B, Chen X, Xie J, Cheng N. Overexpression of Aurora-a enhances invasion and matrix metalloproteinase-2 expression in esophageal squamous cell carcinoma cells. Mol Cancer Res. 2012;10(5):588–96. https://doi.org/10.1158/1541-7786.MCR-11-0416.
Leontovich AA, Jalalirad M, Salisbury JL, Mills L, Haddox C, Schroeder M, et al. NOTCH3 expression is linked to breast cancer seeding and distant metastasis. Breast Cancer Res. 2018;20(1):105. https://doi.org/10.1186/s13058-018-1020-0.
Seniski GG, Camargo AA, Ierardi DF, Ramos EA, Grochoski M, Ribeiro ES, et al. ADAM33 gene silencing by promoter hypermethylation as a molecular marker in breast invasive lobular carcinoma. BMC Cancer. 2009;9(1):80. https://doi.org/10.1186/1471-2407-9-80.
Manica GC, Ribeiro CF, Oliveira MA, Pereira IT, Chequin A, Ramos EA, et al. Down regulation of ADAM33 as a predictive biomarker of aggressive breast Cancer. Sci Rep. 2017;7(1):44414. https://doi.org/10.1038/srep44414.
Xu W, Wang Y, Wang Y, Lv S, Xu X, Dong X. Screening of differentially expressed genes and identification of NUF2 as a prognostic marker in breast cancer. Int J Mol Med. 2019;44(2):390–404. https://doi.org/10.3892/ijmm.2019.4239.
Zhai X, Yang Z, Liu X, Dong Z, Zhou D. Identification of NUF2 and FAM83D as potential biomarkers in triple-negative breast cancer. PeerJ. 2020;8:e9975. https://doi.org/10.7717/peerj.9975.
Chen C, Chen S, Pang L, Yan H, Luo M, Zhao Q, et al. Analysis of the expression of cell division cycle-associated genes and its prognostic significance in human lung carcinoma: a review of the literature databases. Biomed Res Int. 2020;2020:6412593.
Wu ZH, Fang M, Zhou Y. Comprehensive analysis of the expression and prognosis for CDCAs in head and neck squamous cell carcinoma. PLoS One. 2020;15(7):e0236678. https://doi.org/10.1371/journal.pone.0236678.
Phan NN, Wang CY, Li KL, Chen CF, Chiao CC, Yu HG, et al. Distinct expression of CDCA3, CDCA5, and CDCA8 leads to shorter relapse free survival in breast cancer patient. Oncotarget. 2018;9(6):6977–92. https://doi.org/10.18632/oncotarget.24059.
Blanco G, Holli K, Heikkinen M, Kallioniemi OP, Taskinen P. Prognostic factors in recurrent breast cancer: relationships to site of recurrence, disease-free interval, female sex steroid receptors, ploidy and histological malignancy grading. Br J Cancer. 1990;62(1):142–6. https://doi.org/10.1038/bjc.1990.247.
Cao Y, Zhang G, Bao H, Zhang S, Zhang J, Zhao Z, et al. Development of a dual-energy spectral CT based nomogram for the preoperative discrimination of mutated and wild-type KRAS in patients with colorectal cancer. Clin Imaging. 2021;69:205–12. https://doi.org/10.1016/j.clinimag.2020.08.023.
He S, Wang W, Wan Z, Shen H, Zhao Y, You Z, et al. FAM83B inhibits ovarian cancer cisplatin resistance through inhibiting Wnt pathway. Oncogenesis. 2021;10(1):6. https://doi.org/10.1038/s41389-020-00301-y.
Cipriano R, Graham J, Miskimen KL, Bryson BL, Bruntz RC, Scott SA, et al. FAM83B mediates EGFR- and RAS-driven oncogenic transformation. J Clin Invest. 2012;122(9):3197–210. https://doi.org/10.1172/JCI60517.
Cipriano R, Miskimen KL, Bryson BL, Foy CR, Bartel CA, Jackson MW. FAM83B-mediated activation of PI3K/AKT and MAPK signaling cooperates to promote epithelial cell transformation and resistance to targeted therapies. Oncotarget. 2013;4(5):729–38. https://doi.org/10.18632/oncotarget.1027.
Shen CQ, Yan TT, Liu W, Zhu XQ, Tian XL, Fu XL, et al. High expression of FAM83B predicts poor prognosis in patients with pancreatic ductal adenocarcinoma and correlates with cell cycle and cell proliferation. J Cancer. 2017;8(16):3154–65. https://doi.org/10.7150/jca.20086.
Okabe N, Ezaki J, Yamaura T, Muto S, Osugi J, Tamura H, et al. FAM83B is a novel biomarker for diagnosis and prognosis of lung squamous cell carcinoma. Int J Oncol. 2015;46(3):999–1006. https://doi.org/10.3892/ijo.2015.2817.
Le KN, Gibiansky L, Good J, Davancaze T, van Lookeren CM, Loyet KM, et al. A mechanistic pharmacokinetic/pharmacodynamic model of factor D inhibition in cynomolgus monkeys by lampalizumab for the treatment of geographic atrophy. J Pharmacol Exp Ther. 2015;355(2):288–96. https://doi.org/10.1124/jpet.115.227223.
Loveridge CJ, Slater S, Campbell KJ, Nam NA, Knight J, Ahmad I, et al. BRF1 accelerates prostate tumourigenesis and perturbs immune infiltration. Oncogene. 2020;39(8):1797–806. https://doi.org/10.1038/s41388-019-1106-x.
Barnum SR, Ishii Y, Agrawal A, Volanakis JE. Production and interferon-gamma-mediated regulation of complement component C2 and factors B and D by the astroglioma cell line U105-MG. Biochem J. 1992;287(Pt 2):595–601.
Necchi A, Eigl BJ, Yang ES, Bae S, Chandrashekar D, Chen D, et al. Gene expression profiling of advanced penile squamous cell carcinoma receiving Cisplatin-based chemotherapy improves prognostication and identifies potential therapeutic targets. Eur Urol Focus. 2018;4(5):733–6. https://doi.org/10.1016/j.euf.2016.08.001.
Zhang K, Liu J, Li C, Peng X, Li H, Li Z. Identification and validation of potential target genes in papillary thyroid cancer. Eur J Pharmacol. 2019;843:217–25. https://doi.org/10.1016/j.ejphar.2018.11.026.
Lacher MD, Bauer G, Fury B, Graeve S, Fledderman EL, Petrie TD, et al. SV-BR-1-GM, a clinically effective GM-CSF-secreting breast Cancer cell line, expresses an immune signature and directly activates CD4(+) T lymphocytes. Front Immunol. 2018;9:776. https://doi.org/10.3389/fimmu.2018.00776.
Cimino-Mathews A, Foote JB, Emens LA, et al. Oncology (Williston Park, NY). 2015;29(5):375–85.
Yen MC, Huang YC, Kan JY, Kuo PL, Hou MF, Hsu YL. S100B expression in breast cancer as a predictive marker for cancer metastasis. Int J Oncol. 2018;52(2):433–40. https://doi.org/10.3892/ijo.2017.4226.
Galbiatti AL, Castro R, Caldas HC, Padovani JA Jr, Pavarino EC, Goloni-Bertollo EM. Alterations in the expression pattern of MTHFR, DHFR, TYMS, and SLC19A1 genes after treatment of laryngeal cancer cells with high and low doses of methotrexate. Tumour Biol. 2013;34(6):3765–71. https://doi.org/10.1007/s13277-013-0960-3.
Le K, Guo H, Zhang Q, Huang X, Xu M, Huang Z, et al. Gene and lncRNA co-expression network analysis reveals novel ceRNA network for triple-negative breast cancer. Sci Rep. 2019;9(1):15122. https://doi.org/10.1038/s41598-019-51626-7.
Chen W, Liao L, Lai H, Yi X, Wang D. Identification of core biomarkers associated with pathogenesis and prognostic outcomes of laryngeal squamous-cell cancer using bioinformatics analysis. Eur Archiv Oto-rhino-laryngology. 2020;277(5):1397–408.
Shi T, Jiang R, Wang P, Xu Y, Yin S, Cheng X, et al. Significant association of the EXO1 rs851797 polymorphism with clinical outcome of ovarian cancer. OncoTargets Ther. 2017;10:4841–51. https://doi.org/10.2147/OTT.S141668.
Zhang M, Zhao D, Yan C, Zhang L, Liang C. Associations between nine polymorphisms in EXO1 and Cancer susceptibility: a systematic review and meta-analysis of 39 case-control studies. Sci Rep. 2016;6(1):29270. https://doi.org/10.1038/srep29270.
Qi L, Zhou B, Chen J, Hu W, Bai R, Ye C, et al. Significant prognostic values of differentially expressed-aberrantly methylated hub genes in breast cancer. J Cancer. 2019;10(26):6618–34. https://doi.org/10.7150/jca.33433.
Franzese C, Di Brina L, D'Agostino G, Franceschini D, Comito T, De Rose F, et al. Predictive factors for survival outcomes of oligometastatic prostate cancer patients treated with metastases-directed therapy: a recursive partitioning-based analysis. J Cancer Res Clin Oncol. 2019;145(10):2469–79. https://doi.org/10.1007/s00432-019-03007-w.
Jiang Y, Zhang M, Qian Y, Xu E, Zhang J, Chen X. Rbm24, an RNA-binding protein and a target of p53, regulates p21 expression via mRNA stability. J Biol Chem. 2014;289(6):3164–75. https://doi.org/10.1074/jbc.M113.524413.
Xu E, Zhang J, Zhang M, Jiang Y, Cho SJ, Chen X. RNA-binding protein RBM24 regulates p63 expression via mRNA stability. Mol Cancer Res. 2014;12(3):359–69. https://doi.org/10.1158/1541-7786.MCR-13-0526.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from National Natural Sciences Foundation of China (81873219, 81774180), Natural Science Foundation of Jiangsu Province (BK20181425), Young Elite Scientists Sponsorship Program by CACM (CACM-2018-QNRC2-B07), National Innovation and Entrepreneurship training Program for College Students (201910315006). In addition, this work was also financially supported in part by the grants from a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Contributions
Lei Bi designed the study. Yiduo Liu, Linxin Teng and Lei Bi contributed to the writing of the manuscript. Linxin Teng and Yiduo Liu performed the data analysis. Shiyi Fu, Haodi Wang and Zhengjun Li revised the images. Yiduo Liu, Guiyang Wang and Chao Ding performed the literature search and collected data for the manuscript. Lei Bi provided manuscript revision, scientific research fund declaration and fund support. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
No potential conflicts of interest were disclosed.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Statistics of microarray datasets from GEO and TCGA.
Additional file 2: Table S2.
The DEGs in GEO and TCGA datasets.
Additional file 3: Table S3.
ROC analysis of DEGs based on TCGA samples.
Additional file 4: Table S4.
Related information of all genes involved in GO and KEGG analysis.
Additional file 5: Table S5.
Related information of all genes involved in univariate Cox regression analysis.
Additional file 6: Table S6.
The clinical data and the result of Cox regression analysis in DFI and PFI models.
Additional file 7: Figure S1.
Survival analysis of TNBC subtypes.
Additional file 8: Figure S2.
Scatter plots of correlation analysis of prognostic factors.
Additional file 9: Table S7.
OS analysis of TNBC and Breast cancer.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, Y., Teng, L., Fu, S. et al. Highly heterogeneous-related genes of triple-negative breast cancer: potential diagnostic and prognostic biomarkers. BMC Cancer 21, 644 (2021). https://doi.org/10.1186/s12885-021-08318-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08318-1