
Abstract
Background
Numerous studies have highlighted that long non-coding RNAs (lncRNAs) can bind to microRNA (miRNA) sites as competing endogenous RNAs (ceRNAs), thereby affecting and regulating the expression of mRNAs and target genes. These lncRNA-associated ceRNAs have been theorized to play a significant role in cancer initiation and progression. However, the roles and functions of the lncRNA-miRNA-mRNA ceRNA network in squamous cell carcinoma of the tongue (SCCT) are still unclear.
Methods
The miRNA, mRNA and lncRNA expression profiles from 138 patients with SCCT were downloaded from The Cancer Genome Atlas database. We identified the differential expression of miRNAs, mRNAs, and lncRNAs using the limma package of R software. We used the clusterProfiler package for GO and KEGG pathway annotations. The survival package was used to estimate survival analysis according to the Kaplan-Meier curve. Finally, the GDCRNATools package was used to construct the lncRNA-miRNA-mRNA ceRNA network.
Results
In total, 1943 SCCT-specific mRNAs, 107 lncRNAs and 100 miRNAs were explored. Ten mRNAs (CSRP2, CKS2, ADGRG6, MB21D1, GMNN, RIPOR3, RAD51, PCLAF, ORC1, NAGS), 9 lncRNAs (LINC02560, HOXC13 − AS, FOXD2 − AS1, AC105277.1, AC099850.3, STARD4 − AS1, SLC16A1 − AS1, MIR503HG, MIR100HG) and 8 miRNAs (miR − 654, miR − 503, miR − 450a, miR − 379, miR − 369, miR − 190a, miR − 101, and let−7c) were found to be significantly associated with overall survival (log-rank p < 0.05). Based on the analysis of the lncRNA-miRNA-mRNA ceRNA network, one differentially expressed (DE) lncRNA, five DEmiRNAs, and three DEmRNAs were demonstrated to be related to the pathogenesis of SCCT.
Conclusions
In this study, we described the gene regulation by the lncRNA-miRNA-mRNA ceRNA network in the progression of SCCT. We propose a new lncRNA-associated ceRNA that could help in the diagnosis and treatment of SCCT.
Similar content being viewed by others

Background
Head and neck squamous cell carcinoma (HNSCC), which is a disease that causes serious harm to humans, is highly correlated with alcohol consumption, tobacco smoking, and betel nut chewing, and human papillomavirus infection. Squamous cell carcinoma of the tongue (SCCT) is a particular subtype and the main cause of patient mortality and morbidity from HNSCC [1, 2]. In general, the clinical features and treatment strategies for SCCT are similar to those of other HNSCCs, with surgical resection being the primary treatment choice. However, due to late diagnosis of locally advanced malignancies, in many cases of SCCT, surgery is either no longer an option, or should be avoided to maintain the patient’s quality of life [3, 4]. Despite the advances in treatment options, the prognosis of patients with advanced SCCT remains poor [5]. In China, although pingyangmycin and/or cisplatin-based chemotherapies have shown good results, chemotherapy resistance always develops later and causes the therapy to fail [6]. In the past three decades, the 5-year survival rate of patients with SCCT was less than 50% [7]. Therefore, the main goal of our research has been to obtain more knowledge about SCCT cells and to identify novel therapeutic targets for treating the disease.
Long non-coding RNAs (lncRNAs), which do not have protein-coding functions, have recently attracted increasing research attention [8, 9]. These RNAs play a significant role in different cellular processes, particularly in numerous kinds of tumors [10,11,12]. For example, lncRNAs can act as biomarkers for the prognosis and diagnosis of lung adenocarcinoma [13]. MicroRNAs (miRNAs) are small, endogenous, non-coding RNAs composed of 19–25 nucleotides [14, 15]. They exert the important function of regulating gene expression, and their regulatory networks are involved in many biological processes [16,17,18]. In 2011, Salmena et al. proposed the competitive endogenous RNA (ceRNA) hypothesis [19], which was subsequently supported by several lines of evidence [20,21,22,23,24]. This hypothesis describes the competitive activity of some RNAs (as ceRNAs) for common binding sites of target miRNAs, thereby altering the function of the target miRNA [25]. The core concept is that ceRNAs interact with target miRNAs through miRNA response elements to control the transcriptome on a large scale. In the past several years, lncRNAs and SCCT were confirmed to be closely related. For instance, expression of the lncRNA SNHG6 is significantly increased in tongue cancer, and interference with SNHG6 expression can inhibit the proliferation and epithelial–mesenchymal transition (EMT) of tongue cancer cells [26]. Zhang et al. found that the oncogenic lncRNA KCNQ1OT1 plays a vital role in SCCT growth and chemoresistance, and can be used as a new target for SCCT treatment [27]. However, previous studies had focused on the mechanism of a single lncRNA-miRNA-mRNA axis, and there is currently no reported ceRNA network in SCCT. Consequently, it is extremely important to investigate the role of ceRNA networks in the poor prognosis of SCCT. By further learning how lncRNAs function in the pathogenesis of SCCT, we may find solutions to the most pressing challenges faced in treating this disease.
In this study, the mRNA, miRNA, and lncRNA expression profiles of SCCT and normal tissues were downloaded from The Cancer Genome Atlas (TCGA). In addition, through comprehensive analysis, the ceRNA network for SCCT was builted, which will serve to find new targets and pathways for the development of treatments to prolong patient survival times. Finally, we conducted a prognostic analysis with several important lncRNAs and found a biomarker that could predict survival in patients with SCCT.
Methods
Patients and samples
The SCCT cases data of clinical and RNA expression were collected from TCGA database. The exclusion criteria were including: (i) histological diagnosis was not SCCT; (ii) no complete data (including gender, age, survival status, stage, and survival time) for analysis [28]. 118 SCCT patients were enrolled in the study. The number of patients aged < 68 years was 49, 69 patients were ≥ 68 years old. 43 patients were female and 75 patients were male. The number of stage I, II, III, IVa and IVb patients were 13, 20, 27, 56 and 2, respectively. The number of patients, who were white, Asian, black or African American and not available, were 106, 5, 5 and 2, respectively. The number of patients, who were hispanic or latino, were 104. 9 patients were not hispanic or latino, and 5 patients were not reported. 46 patients were dead, and 72 patients were alive. SCCT characteristics and clinical data of the patients are showed in Table 1 and Additional file 3: Table S1.
RNA sequence analysis
RNA expression data of SCCT patients were available from TCGA database. The raw reads of lncRNA and mRNA data were post-treated and normalized in R software (Additional file 1: Figure S1). The miRNA expression data from TCGA database were normalized in R software (Additional file 2: Figure S2). The tumor tissue and adjacent non-tumor tissue of SCCT patients were facilitated differential expressions of mRNA, lncRNA, and miRNA. Furthermore, intersection of lncRNA, miRNA and mRNA was selected [13].
Differentially expressed analysis
Compared to the normal group with SCCT, “limma” package in R software was used to identify the differentially expressed mRNAs (DEmRNAs) with thresholds of |fold Change (FC)| > 2.0 and P value < 0.01 and differentially expressed miRNAs with |FC| > 2.5 and P value < 0.01.
Functional enrichment analysis
“ClusterProfiler” package in R software was used for functional enrichment analysis, and GO biological processes and KEGG pathways at the significant level (q-value < 0.01) were employed.
Survival analysis
To determine the prognostic characteristics of DERNAs, combining the clinical data the survival curves of these samples with differentially expressed mRNA, lncRNA and miRNA were plotted by using the “survival” package in R based on Kaplan-Meier curve analysis. P values < 0.05 were regarded as significant.
Construction of lncRNA-miRNA-mRNA ceRNA network
The lncRNA-miRNA-mRNA ceRNA network was based on the theory that lncRNAs can directly interact by invoking miRNA sponges to regulate mRNA activity [29]. “GDCRNATools” (http://bioconductor.org/packages/devel/bioc/html/GDCRNATools.html) package in R software were used to establish ceRNA network [30]. The ceRNA network was plotted with Cytoscape v3.6.0 [31]. The plug-in BinGO of Cytoscape is an APP for BF network of the hub genes [32].
Results
Identification of differentially expressed lncRNA, miRNA and mRNA
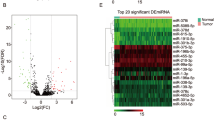
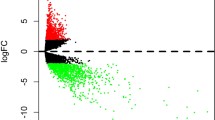
We explored 1943 SCCT-specific mRNAs (1007 downregulated and 936 upregulated; Table 2 and Fig. 1) and 107 lncRNAs (34 downregulated and 73 upregulated; Fig. 1, Table 2, and Table 3). The differentially expressed genes (DEGs) are shown in Fig. 2a. Additionally, 100 miRNAs (44 upregulated and 56 downregulated; Fig. 2b, and Table 4) were found.

Column diagram of DEGs
DEGs were selected with thresholds of fold change > 2 and p < 0.01.

Volcano Plot of DEGs and DEmRNAs. a Volcano Plot of DEGs. b Volcano Plot of differentially expressed miRNA. Upregulated genes are marked in light red; downregulated genes are marked in light green. (DEGs were selected with thresholds of fold change > 2 and p < 0.01, DEmRNAs were selected with thresholds of fold change > 2.5 and p < 0.01)
GO and pathway analysis of DEGs
GO analysis results showed that changes in biological processes (BP) of DEGs were significantly enriched in extracellular structure organization, extracellular matrix organization, urogenital system development, muscle contraction, collagen metabolic process, mitotic nuclear division, renal system development, collagen catabolic process, sister chromatid segregation, and collagen metabolic process (Fig. 3a). Changes in cell component (CC) of DEGs were mainly enriched in proteinaceous extracellular matrix, endoplasmic reticulum lumen, apical part of cell, contractile fiber, myofibril, contractile fiber part, sarcomere, extracellular matrix component, basement membrane, basal lamina (Fig. 3b). Changes in molecular function (MF) were mainly enriched in actin binding, growth factor binding, coenzyme binding, microtubule binding, iron ion binding, glycosaminoglycan binding, collagen binding, structural constituent of muscle, extracellular matrix structural constituent, platelet−derived growth factor binding (Fig. 3c). KEGG pathway analysis revealed that the DEGs were mainly enriched in focal adhesion, human papillomavirus infection, ECM − receptor interaction, protein digestion and absorption, small cell lung cancer, arginine and proline metabolism, PI3K-Akt signaling pathway, dilated cardiomyopathy (DCM), valine, leucine and isoleucine degradation, cell cycle (Fig. 4).

GO enrichment analysis of DEGs in SCCT. (Top 10). a Bubble Plot of BP. b Bubble Plot of CC. c Bubble Plot of MF

Top 10 enrichment of KEGG pathway analysis of DEGs
Survival analysis with the DEGs and DEmRNAs
We studied the association of the DEGs and DEmRNAs with patient’ survival to identify the key genes and mRNAs that were related to the prognosis of patients with SCCT. We identified 10 mRNAs (CSRP2, CKS2, ADGRG6, MB21D1, GMNN, RIPOR3, RAD51, PCLAF, ORC1, NAGS), 9 lncRNAs (LINC02560, HOXC13 − AS, FOXD2 − AS1, AC105277.1, AC099850.3, STARD4 − AS1, SLC16A1 − AS1, MIR503HG, MIR100HG) and 8 miRNAs (miR − 654, miR − 503, miR − 450a, miR − 379, miR − 369, miR − 190a, miR − 101, let−7c) that were significantly differentially expressed in the survival analyses (Fig. 5a-c).

Kaplan-Meier survival curves for mRNAs (a), lncRNAs (b), and miRNAs (c) associated with overall survival (Top 10)
Construction and analysis of the lncRNA-miRNA-mRNA ceRNA network
We built the ceRNA network on the basis of the miRNA, lncRNA, and mRNA the expression profiles in patients with SCCT. In total, 27 miRNA nodes, 53 mRNA nodes, 6 lncRNA nodes, and 152 edges were identified as differentially expressed profiles. The network is showed in Fig. 6. It is well known that lncRNAs and mRNAs have co-expression patterns in ceRNA networks. Thus, we chose a hub lncRNA (degree> 5, Additional file 4: Table S2) and its linked mRNAs and miRNAs in the triple global network and then reconstructed the sub-network. As shown in Fig. 7, the lncRNA KCNQ1OT1-miRNA-mRNA sub-network was composed of 1 lncRNA node, 7 miRNA nodes, 11 mRNA nodes, and 41 edges.

The lncRNA-miRNA-mRNA Competing endogenous RNA network. The rectangles indicate mRNAs in light green, ellipses represent lncRNAs in light purple and diamonds represent miRNAs in light red

The sub-network of lncRNA KCNQ1OT1 and the GO terms interaction network. The lncRNA KCNQ1OT1 sub-network. The rectangles indicate mRNAs in light green, ellipses represent lncRNAs in light purple and diamonds represent miRNAs in light red
Discussion
SCCT, a major type of HNSCC, is a refractory cancer under current therapeutics [33]. Studies have demonstrated that lncRNAs regulate gene expression through a variety of pathways, contributing to tumorigenesis and tumor metastasis [34, 35]. The ceRNA hypothesis proposes a new regulatory mechanism mediated by lncRNAs that are used as endogenous miRNA sponges [19, 36,37,38]. In this study, we found the genes and mRNAs that were differentially expressed between normal and tumor tissue. Through GO and KEGG analyses, we further analyzed the pathways and functions in which the DEGs are involved. The GO biological processes results suggested that specific genes may be concentrated in several process areas, such as extracellular structures, muscle contraction, and mitotic nuclear division. Some of the annotated pathways have been shown to be associated with cancer in previous studies. PI3K-Akt signaling is involved in cell proliferation and growth as well as down-regulating cell apoptosis [39]. Recent preclinical and clinical studies of highly selective agents that target various regulators of the mammalian cell cycle have demonstrated cell-cycle arrest in models of human cancer [40]. Through survival analysis, we identified 10 mRNAs (CSRP2, CKS2, ADGRG6, MB21D1, GMNN, RIPOR3, RAD51, PCLAF, ORC1, NAGS), 9 lncRNAs (LINC02560, HOXC13 − AS, FOXD2 − AS1, AC105277.1, AC099850.3, STARD4 − AS1, SLC16A1 − AS1, MIR503HG, MIR100HG) and 8 miRNAs (miR − 654, miR − 503, miR − 450a, miR − 379, miR − 369, miR − 190a, miR − 101, let−7c) that were significantly related to the overall survival of patients with SCCT. Next, by using bioinformatics tools, we builted a ceRNA network with SCCT-specific miRNA and lncRNA expression and selected the hub lncRNA KCNQ1OT1 to construct a sub-network.
KCNQ1OT1, also known as KCNQ1 overlapping transcript 1, is an imprinted antisense lncRNA in the KCNQ1 locus [41, 42]. Early studies have shown that KCNQ1OT1 is up-regulated and involved in the tumorigenesis of breast cancer and hepatocellular carcinoma [43, 44]. Zhang et al. found that KCNQ1OT1 could induce SCCT cell growth and inhibit the sensitivity of the tumor to cisplatin [27]. Previous studies have shown that KCNQ1OT1 acts as an oncogene and plays a key role in promoting SCCT cell growth and chemotherapy resistance.
Conclusion
We constructed a SCCT-specific ceRNA network and chose a hub lncRNA for SCCT by bioinformatics analysis. To the best of our knowledge, only a limited number of studies have analyzed lncRNA obtained from large-scale samples. We provide a method for identifying potential lncRNA biomarkers. Furthermore, we found the ceRNA network in SCCT, which should help further our understanding of the mechanism underlying the pathogenesis of this disease.
Availability of data and materials
All relevant data are within the manuscript.
Abbreviations
- BP:
-
Biological processes
- CC:
-
Cell component
- ceRNA:
-
Competing endogenous RNA
- DCM:
-
Dilated cardiomyopathy
- DEGs:
-
Differentially expressed genes
- EMT:
-
Epithelial–mesenchymal transition
- GO:
-
Gene Ontology
- HNSCC:
-
Head and neck squamous cell carcinoma
- HPV:
-
Human papillomavirus
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- lncRNA:
-
long non-coding RNA
- MF:
-
Molecular function
- miRNAs:
-
microRNAs
MREs
MicroRNA response elements
- ncRNA:
-
non-coding RNA
- SCCT:
-
Squamous cell carcinoma of tongue
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
Murphy CT, Galloway TJ, Handorf EA, Egleston BL, Wang LS, Mehra R, Flieder DB, Ridge JA. Survival impact of increasing time to treatment initiation for patients with head and neck Cancer in the United States. J Clin Oncol. 2016;34(2):169–78.
Adelstein DJ, Li Y, Adams GL, Wagner H Jr, Kish JA, Ensley JF, Schuller DE, Forastiere AA. An intergroup phase III comparison of standard radiation therapy and two schedules of concurrent chemoradiotherapy in patients with unresectable squamous cell head and neck cancer. J Clin Oncol. 2003;21(1):92–8.
Al-Sarraf M, Pajak TF, Marcial VA, Mowry P, Cooper JS, Stetz J, Ensley JF, Velez-Garcia E. Concurrent radiotherapy and chemotherapy with cisplatin in inoperable squamous cell carcinoma of the head and neck. An RTOG Study. Cancer. 1987;59(2):259–65.
Chinn SB, Myers JN. Oral cavity carcinoma: current management, controversies, and future directions. J Clin Oncol. 2015;33(29):3269–76.
Mascitti M, Orsini G, Tosco V, Monterubbianesi R, Balercia A, Putignano A, Procaccini M, Santarelli A. An overview on current non-invasive diagnostic devices in Oral oncology. Front Physiol. 2018;9:1510.
Gao L, Wang FQ, Li HM, Yang JG, Ren JG, He KF, Liu B, Zhang W, Zhao YF. CCL2/EGF positive feedback loop between cancer cells and macrophages promotes cell migration and invasion in head and neck squamous cell carcinoma. Oncotarget. 2016;7(52):87037–51.
Mendell JT. Targeting a long noncoding RNA in breast Cancer. N Engl J Med. 2016;374(23):2287–9.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62.
Schmitt AM, Chang HY. Long noncoding RNAs in Cancer pathways. Cancer Cell. 2016;29(4):452–63.
Arun G, Diermeier SD, Spector DL. Therapeutic targeting of long non-coding RNAs in Cancer. Trends Mol Med. 2018;24(3):257–77.
Slaby O, Laga R, Sedlacek O. Therapeutic targeting of non-coding RNAs in cancer. Biochem J. 2017;474(24):4219–51.
Sui J, Li YH, Zhang YQ, Li CY, Shen X, Yao WZ, Peng H, Hong WW, Yin LH, Pu YP, et al. Integrated analysis of long non-coding RNAassociated ceRNA network reveals potential lncRNA biomarkers in human lung adenocarcinoma. Int J Oncol. 2016;49(5):2023–36.
Wang X, Chen X, Meng Q, Jing H, Lu H, Yang Y, Cai L, Zhao Y. MiR-181b regulates cisplatin chemosensitivity and metastasis by targeting TGFbetaR1/Smad signaling pathway in NSCLC. Sci Rep. 2015;5:17618.
Wang X, Yin H, Zhang H, Hu J, Lu H, Li C, Cao M, Yan S, Cai L. NF-kappaB-driven improvement of EHD1 contributes to erlotinib resistance in EGFR-mutant lung cancers. Cell Death Dis. 2018;9(4):418.
Zhang H, Li Y, Lai M. The microRNA network and tumor metastasis. Oncogene. 2010;29(7):937–48.
Gennarino VA, D'Angelo G, Dharmalingam G, Fernandez S, Russolillo G, Sanges R, Mutarelli M, Belcastro V, Ballabio A, Verde P, et al. Identification of microRNA-regulated gene networks by expression analysis of target genes. Genome Res. 2012;22(6):1163–72.
Li L, Wang D, Xue M, Mi X, Liang Y, Wang P. UTR shortening identifies high-risk cancers with targeted dysregulation of the ceRNA network. Sci Rep. 2014;4(3):5406.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell. 2011;146(3):353–8.
Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4(9):721–6.
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465(7301):1033–8.
Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y, Chen N, Sun F, Fan Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010;38(16):5366–83.
Lee DY, Jeyapalan Z, Fang L, Yang J, Zhang Y, Yee AY, Li M, Du WW, Shatseva T, Yang BB. Expression of versican 3′-untranslated region modulates endogenous microRNA functions. PLoS One. 2010;5(10):e13599.
Jeyapalan Z, Deng Z, Shatseva T, Fang L, He C, Yang BB. Expression of CD44 3′-untranslated region regulates endogenous microRNA functions in tumorigenesis and angiogenesis. Nucleic Acids Res. 2011;39(8):3026–41.
Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–52.
Zhao Y, Wang J, Ma K. Knockdown of lncRNA SNHG6 inhibites the proliferation and epithelial mesenchymal transition in tongue cancer cells. Xi bao yu fen zi mian yi xue za zhi. 2018;34(9):806–11.
Zhang S, Ma H, Zhang D, Xie S, Wang W, Li Q, Lin Z, Wang Y. LncRNA KCNQ1OT1 regulates proliferation and cisplatin resistance in tongue cancer via miR-211-5p mediated Ezrin/Fak/Src signaling. Cell Death Dis. 2018;9(7):742.
Yin H, Wang X, Zhang X, Wang Y, Zeng Y, Xiong Y, Li T, Lin R, Zhou Q, Ling H, et al. Integrated analysis of long noncoding RNA associated-competing endogenous RNA as prognostic biomarkers in clear cell renal carcinoma. Cancer Sci. 2018;109(10):3336–49.
Guo LL, Song CH, Wang P, Dai LP, Zhang JY, Wang KJ. Competing endogenous RNA networks and gastric cancer. World J Gastroenterol. 2015;21(41):11680–7.
Li R, Qu H, Wang S, Wei J, Zhang L, Ma R, Lu J, Zhu J, Zhong WD, Jia Z. GDCRNATools: an R/Bioconductor package for integrative analysis of lncRNA, miRNA and mRNA data in GDC. Bioinformatics. 2018;34(14):2515–7.
Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–2.
Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–9.
Otsuka Y, Sato H, Oikawa T, Onodera Y, Nam JM, Hashimoto A, Fukunaga K, Hatanaka KC, Hatanaka Y, Matsuno Y, et al. High expression of EPB41L5, an integral component of the Arf6-driven mesenchymal program, correlates with poor prognosis of squamous cell carcinoma of the tongue. Cell Commun Signal. 2016;14(1):28.
Tripathi V, Shen Z, Chakraborty A, Giri S, Freier SM, Wu X, Zhang Y, Gorospe M, Prasanth SG, Lal A, et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013;9(3):e1003368.
Ying L, Huang Y, Chen H, Wang Y, Xia L, Chen Y, Liu Y, Qiu F. Downregulated MEG3 activates autophagy and increases cell proliferation in bladder cancer. Mol BioSyst. 2013;9(3):407–11.
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147(2):358–69.
Wang Y, Xu Z, Jiang J, Xu C, Kang J, Xiao L, Wu M, Xiong J, Guo X, Liu H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell. 2013;25(1):69–80.
Kallen AN, Zhou XB, Xu J, Qiao C, Ma J, Yan L, Lu L, Liu C, Yi JS, Zhang H, et al. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell. 2013;52(1):101–12.
Danielsen SA, Eide PW, Nesbakken A, Guren T, Leithe E, Lothe RA. Portrait of the PI3K/AKT pathway in colorectal cancer. Biochim Biophys Acta. 2015;1855(1):104–21.
Mills CC, Kolb EA, Sampson VB. Recent advances of cell-cycle inhibitor therapies for pediatric Cancer. Cancer Res. 2017;77(23):6489–98.
Zhang H, Zeitz MJ, Wang H, Niu B, Ge S, Li W, Cui J, Wang G, Qian G, Higgins MJ, et al. Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus. J Cell Biol. 2014;204(1):61–75.
Kanduri C. Kcnq1ot1: a chromatin regulatory RNA. Semin Cell Dev Biol. 2011;22(4):343–50.
Zhang Z, Weaver DL, Olsen D, deKay J, Peng Z, Ashikaga T, Evans MF. Long non-coding RNA chromogenic in situ hybridisation signal pattern correlation with breast tumour pathology. J Clin Pathol. 2016;69(1):76–81.
Wan J, Huang M, Zhao H, Wang C, Zhao X, Jiang X, Bian S, He Y, Gao Y. A novel tetranucleotide repeat polymorphism within KCNQ1OT1 confers risk for hepatocellular carcinoma. DNA Cell Biol. 2013;32(11):628–34.
Acknowledgements
Not applicable.
Funding
This work was supported by High-level University Construction of Guangzhou University of Chinese Medicine (A1-AFD018181A29 and A1–2606–18-414-016). The funding bodies were not involved in the design of this study or in the collection of data, analysis, and interpretation of hereof.
Author information
Authors and Affiliations
Contributions
RSZ, EXZ, QFS and ZJY conducted experiments, collected the data and wrote the manuscript. JWL and DHZ conducted experiments. YT, QFS and ZJY collected data, contributed to the discussion and reviewed the manuscript. YT is the guarantor of this work and, as such, has full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No ethics approval was required for this work. All utilized public data sets were generated by others who obtained ethical approval.
Consent for publication
Not Applicable.
Competing interests
The authors have declared that they have no competing of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Boxplot of normalized RNA expression data (PDF 96 kb)
Additional file 2:
Figure S2. Boxplot of normalized miRNA expression data (PDF 20 kb)
Additional file 3:
Table S1. 118 SCCT patients clinical data (DOCX 25 kb)
Additional file 4:
Table S2. The degree of ceRNA network. (DOCX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, RS., Zhang, EX., Sun, QF. et al. Integrated analysis of lncRNA-miRNA-mRNA ceRNA network in squamous cell carcinoma of tongue. BMC Cancer 19, 779 (2019). https://doi.org/10.1186/s12885-019-5983-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-019-5983-8