
Abstract
In many types of solid tumours, the aberrant expression of the cell adhesion molecule N-cadherin is a hallmark of epithelial-to-mesenchymal transition, resulting in the acquisition of an aggressive tumour phenotype. This transition endows tumour cells with the capacity to escape from the confines of the primary tumour and metastasise to secondary sites. In this review, we will discuss how N-cadherin actively promotes the metastatic behaviour of tumour cells, including its involvement in critical signalling pathways which mediate these events. In addition, we will explore the emerging role of N-cadherin in haematological malignancies, including bone marrow homing and microenvironmental protection to anti-cancer agents. Finally, we will discuss the evidence that N-cadherin may be a viable therapeutic target to inhibit cancer metastasis and increase tumour cell sensitivity to existing anti-cancer therapies.
Similar content being viewed by others


Background
Cancer metastasis is a leading cause of cancer-related mortality. The metastasis of cancer cells within primary tumours is characterised by localised invasion into the surrounding microenvironment, entry into the vasculature and subsequent spread to permissive distant organs [1, 2]. In many epithelial cancers, metastasis is facilitated by the genetic reprogramming and transitioning of cancer cells from a non-motile, epithelial phenotype into a migratory, mesenchymal-like phenotype, a process known as epithelial-to-mesenchymal transition (EMT) [3, 4]. A common feature of EMT is the loss of epithelial cadherin (E-cadherin) expression and the concomitant up-regulation or de novo expression of neural cadherin (N-cadherin). This so-called “cadherin switch” is associated with increased migratory and invasive behaviour [5, 6] and inferior patient prognosis [7,8,9,10]. A major consequence of E-cadherin down-regulation is the loss of stable epithelial cell-cell adhesive junctions, apico-basal cell polarity and epithelial tissue structure, thereby facilitating the release of cancer cells from the primary tumour site [11, 12]. In contrast to the migration-suppressive role of E-cadherin, N-cadherin endows tumour cells with enhanced migratory and invasive capacity, irrespective of E-cadherin expression [13]. Thus, the acquisition of N-cadherin appears to be a critical step in epithelial cancer metastasis and disease progression.
In this review, we will discuss how N-cadherin promotes the metastatic behaviour of tumour cells by directly mediating cell-cell adhesion, and by its involvement in modulating critical signalling pathways implicated in metastatic events. In addition, we will discuss the emerging relevance of N-cadherin in haematological malignancies, namely leukaemias and multiple myeloma. Finally, we will review the emerging evidence that N-cadherin may be a viable therapeutic target to inhibit cancer metastasis and overcome resistance to anti-cancer agents.
Structure and formation of the N-cadherin adhesive complex
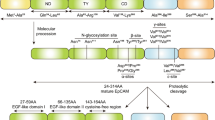
N-cadherin is a member of the calcium-dependent adhesion molecule family of classical cadherins which directly mediate homotypic and heterotypic cell-cell adhesion. N-cadherin is a classical type I cadherin consisting of 5 extracellular domains linked to a functional intracellular domain. The engagement between N-cadherin monomers on opposing cells occurs by reciprocal insertion of a tryptophan residue side-chain on its first extracellular domain (EC1) into the hydrophobic pocket of the partner N-cadherin EC1 (trans adhesion). In addition, the stabilisation of N-cadherin-mediated adhesion requires the clustering of adjacent monomers on the surface of the same cell, involving the His-Ala-Val (HAV) motif on EC1 and a recognition sequence on the second extracellular domain (EC2) of the lateral N-cadherin monomer (cis adhesion) [14,15,16]. The membrane expression and lateral clustering of N-cadherin is dependent upon p120 catenin, which localises N-cadherin at cholesterol-rich microdomains [17, 18]. The initial ligation of N-cadherin extracellular domains triggers the activation of the Rho GTPase family member Rac, which stimulates localised actin filament assembly and the formation of membrane protrusions at points of cell-cell contact [19, 20]. The subsequent activation of the Rho GTPase family member RhoA, at the expense of Rac function, facilitates the maturation of N-cadherin-based cell-cell junctions by triggering the sequestration of β-catenin to the cadherin intracellular domain [21, 22]. β-catenin serves as a critical link to α-catenin which accumulates at nascent cell-cell junctions and suppresses actin branching. In addition, α-catenin facilitates the anchorage of the N-cadherin-catenin complex to the actin cytoskeleton via actin-binding proteins such as cortactin and α-actinin, thereby promoting the maturation of cell-cell contacts [23, 24] (Fig. 1). Notably, the adhesive function of N-cadherin is regulated by post-translational modifications of the N-cadherin-catenin complex. For instance, the stability of the N-cadherin-catenin complex is highly dependent on the phosphorylation status of N-cadherin and the associated catenins, which is regulated by tyrosine kinases, such as Fer and Src, and the tyrosine phosphatase PTP1B [25, 26]. In addition, branched N-glycosylation of N-cadherin EC2 and third extracellular domain regulates N-cadherin-dependent cell adhesion, at least in part, by controlling the lateral clustering of N-cadherin monomers [27].

Schematic representation of the N-cadherin-catenin adhesive complex. The extracellular domains of N-cadherin monomers engage in trans and cis interactions with partner monomers, facilitated by p120-catenin (p120), resulting in a lattice-like arrangement. Interaction between monomers on opposing cells occurs via a reciprocal insertion of tryptophan side-chains (W) on the first extracellular domain (EC1) (trans adhesion). Clustering of N-cadherin monomers on the same cell occurs via a His-Ala-Val (HAV) adhesion motif on EC1 and a recognition sequence on the second extracellular domain (EC2) of the partner monomer (cis adhesion) (inset). Activation of RhoA sequesters β-catenin (β-cat) and results in accumulation of α-catenin (α-cat) to the N-cadherin intracellular domain. This promotes anchorage of the N-cadherin-catenin complex to the actin cytoskeleton via actin-binding proteins, thereby stabilising cell-cell contacts. Initial ligation of N-cadherin extracellular domains also triggers PI3K/Akt signalling which inactivates the pro-apoptotic protein Bad, resulting in activation of the anti-apoptotic protein Bcl-2
The functional role of N-cadherin in solid tumour metastasis
N-cadherin expression is spatiotemporally regulated throughout development and adulthood. In development, N-cadherin plays an important role in morphogenetic processes during the formation of cardiac and neural tissues, and is involved in osteogenesis, skeletal myogenesis and maturation of the vasculature [28,29,30,31,32]. In adulthood, N-cadherin is expressed by numerous cell types including neural cells, endothelial cells, stromal cells and osteoblasts, and is integral to synapse function, vascular stability and bone homeostasis [30, 33,34,35,36]. While N-cadherin is typically absent or expressed at low levels in normal epithelial cells, the aberrant expression of N-cadherin in epithelial cancer cells is a well-documented feature of epithelial malignancies, such as breast, prostate, urothelial and pancreatic cancer, and is associated with disease progression [37,38,39,40]. In a similar manner, the up-regulation of N-cadherin expression is a feature of melanoma progression [41,42,43]. Whilst the aberrant expression of N-cadherin in epithelial tissues is not considered to be oncogenic, or a promoter of solid tumour growth [44,45,46], increased expression of N-cadherin in cancer is widely associated with tumour aggressiveness. Indeed, many studies have demonstrated a significant correlation between elevated N-cadherin levels in epithelial, and some non-epithelial solid tumours, and clinicopathologic features such as increased localised tumour invasion and distant metastasis, and inferior patient prognosis [7, 8, 47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81] (Table 1). Multivariate analyses have also identified that elevated N-cadherin expression is independently associated with inferior patient prognosis in several epithelial malignancies including prostate, lung and bladder cancer [8, 55, 56, 60, 62, 63, 67, 72, 78, 80] (Table 1). The aggressive phenotype and inferior prognosis associated with up-regulated N-cadherin expression in solid tumours is also supported by a recent meta-analysis incorporating patients with various epithelial malignancies [82].
Beyond the prognostic implications of aberrant N-cadherin expression, the relationship between N-cadherin and metastasis is not merely associative. Indeed, there is a wealth of evidence that increased N-cadherin expression enhances the migratory and invasive capacity of multiple epithelial cancer cell types in vitro [83,84,85,86,87]. The ability of N-cadherin to promote epithelial tumour metastasis in vivo was initially demonstrated using the MCF-7 breast cancer cell line, following injection into the mammary fat pad of nude mice. In contrast to wild-type cells, MCF-7 cells ectopically expressing N-cadherin formed tumour metastases in several organs including the liver, pancreas and lymph nodes [88]. Similarly, N-cadherin expression in the mammary epithelium in the transgenic MMTV-PyMT murine breast cancer model resulted in a three-fold increase in the number of pulmonary metastatic foci without affecting the onset or growth of the primary tumour [45]. Using an orthotopic mouse model of pancreatic cancer, the over-expression of N-cadherin in BxPC-3 cells increased the formation of disseminated tumour nodules throughout the abdominal cavity and induced the formation of N-cadherin-expressing lung micro-metastases [85]. Consistent with these findings, enforced expression of N-cadherin in androgen-responsive prostate cancer cells promoted invasion of underlying muscle and lymph node metastasis following subcutaneous injection in castrated mice [89]. Notably, N-cadherin also potentiates the invasiveness of melanoma cells. To this end, studies have demonstrated that N-cadherin promotes the capacity of melanoma cells to migrate on monolayers of dermal fibroblasts and undergo trans-endothelial migration in vitro [86, 90, 91]. Moreover, N-cadherin silencing has been shown to attenuate the ability of intravenously injected melanoma cells to extravasate and form lung metastases in immunocompromised mice [92].
To appreciate how N-cadherin, a cell adhesion molecule, may actively promote cancer cell migration, it is important to consider that the N-cadherin-catenin complex mediates both cell-cell adhesion and pro-metastatic cell signalling. Moreover, the adhesive function and migration-related signalling capacity of N-cadherin can occur simultaneously, or as antagonistic events, adding further complexity to its role in cancer metastasis. In the following section, we describe three key mechanisms by which N-cadherin has been shown to actively promote the migratory capacity of tumour cells: facilitation of collective cell migration, augmentation of fibroblast growth factor-receptor (FGFR) signalling and modulation of canonical Wnt signalling.
N-cadherin promotes collective cell migration
The migration of cells as sheets, clusters or strands, a process termed collective cell migration, frequently occurs throughout development and in adulthood. For instance, collective cell migration occurs in embryogenesis, during gastrulation and neural crest cell migration, and in adult tissues, during wound healing and angiogenesis [93, 94]. In addition, collective cell migration facilitates the invasion of epithelial cells through the localised tumour host microenvironment, thereby promoting metastasis [95]. During this process, collectively migrating cells maintain physical interconnectivity, collective cell polarity and co-ordinated cytoskeletal activity, resulting in a ‘leader-follower’-type cellular arrangement. This promotes more efficient directional migration, in response to a chemotactic gradient, than that of an individual migrating cell [93, 96]. Adhesive complexes are integral to the co-ordinated behaviour of collectively migrating cells by mediating adhesion, signal transduction and mechanotransduction between adjacent cells [94, 97]. Notably, studies have demonstrated that N-cadherin expression by epithelial cancer cells promotes their capacity for collective migration. For instance, N-cadherin has been shown to promote the ability of lung or ovarian cancer cells to form aggregates and collectively invade three-dimensional (3D) collagen matrices or penetrate peritoneal mesothelium-like cell layers in vitro [87, 98]. Similarly, studies in transformed canine kidney epithelial cells (MDCK cells) have shown that N-cadherin promotes aggregate formation which allows directional collective cell migration in a 3D collagen matrix. In these cells, deletion of the entire N-cadherin intracellular domain, or the β-catenin binding domain alone, resulted in greater individual cell detachment and migration from cell clusters, highlighting the importance of the N-cadherin-actin cytoskeleton interaction in collective cell migration. Moreover, over-expression of an N-cadherin mutant in which the extracellular domain was fused to the anti-binding domain of α-catenin hindered the movement of follower cells, demonstrating that dynamic N-cadherin-actin linkage is required for efficient collective cell migration [99].
In addition to maintaining multi-cellular aggregates of tumour cells, studies in N-cadherin-expressing non-tumour cells have demonstrated that N-cadherin also promotes collective cell migration by polarising Rho-family GTPase signalling (e.g. Rac1 and cdc42), known to co-ordinate cytoskeletal remodelling in collectively migrating cells [100, 101]. For example, models of arterial smooth muscle wound-healing and neural crest migration have shown that the asymmetric distribution of N-cadherin-mediated cell-cell adhesion at the lateral and posterior aspects of leader cells promotes directional cell alignment and increased cdc42 and Rac1 activity and protrusion formation at the free leading cell edge, resulting in enhanced migration [102, 103]. Mechanistically, studies in mouse embryonic fibroblasts have demonstrated that N-cadherin-adhesive complexes at the rear of cells suppress localised integrin-α5 activity, thereby polarising integrin and Rac activity towards the free leading edge of the cell [104]. Indeed, functional inhibition of N-cadherin in transformed mammary cells has been shown to reduce integrin-α5-dependent cell migration on fibronectin in vitro [105]. In a similar manner, silencing of N-cadherin expression in melanoma cells perturbs α2β1-integrin-dependent collagen matrix invasion in vitro [106]. Reciprocally, integrin signalling at focal adhesions has been shown to regulate the ability of HeLa cells to engage in N-cadherin-based connections and to promote collective cell migration [107]. Given that integrins play an important role in the activation of Rho signalling [108, 109], it is plausible that N-cadherin may polarise Rho-family GTPase signalling via intercommunication with integrins, thereby promoting the collective migration of cancer cells (Fig. 2a).

Schematic representation of cell signalling events modulated by increased N-cadherin expression in the context of cell migration. a In addition to mediating cellular aggregation, N-cadherin may facilitate the collective migration of tumour cells by excluding focal adhesions and Rac1 activity, and promoting RhoA activity, at sites of N-cadherin-mediated cell-cell contact. The asymmetric distribution of N-cadherin adhesive complexes polarises integrin function and Rac1 activity towards the free edges of cells, thereby directing focal adhesion and lamellipodia formation away from the cell cluster and promoting cell migration. Similar to Rac1, N-cadherin-mediated cell-cell adhesion promotes cdc42 activity at the free edges of cells, resulting in filipodia formation. b Functional interaction between the extracellular domains of N-cadherin and FGFR-1 potentiates FGF-2-activated FGFR-1 signalling by attenuating ligand-induced receptor internalisation. The resulting augmentation of down-stream MEK/ERK and PI3K/Akt signalling promotes the metastatic behaviour of cancer cells by increasing the production of invasion-facilitating molecules such as matrix metalloproteinases (MMPs). c N-cadherin-mediated adhesive complexes and Wnt/β-catenin signalling are thought to compete for the same cellular pool of β-catenin. While N-cadherin sequesters β-catenin from the nucleus, the N-cadherin adhesive complex provides a reservoir of β-catenin which, upon Wnt activation, becomes available for nuclear translocation and TCF/LEF-mediated gene transcription (e.g. CD44 and MMP genes), resulting in the loss of N-cadherin-mediated cellular adhesion in cancer cells
N-cadherin augments fibroblast growth factor receptor signalling
Functional interaction between the extracellular domains of N-cadherin and receptor-tyrosine kinase FGFRs was first recognised as a mechanism by which N-cadherin promoted axonal outgrowth of rat cerebellar neuronal cells. These studies identified that the fourth extracellular domain of N-cadherin (EC4) trans-activated FGFRs to promote neurite outgrowth independent of FGF ligands, suggesting that N-cadherin can act as a surrogate ligand of FGFRs [33, 110]. The physical interaction of N-cadherin and FGFRs has also been shown in breast and pancreatic cancer cells [111,112,113,114]. Evidence that FGFR plays a functional role in N-cadherin-mediated cancer metastasis has been demonstrated in BT-20 and PyMT breast cancer cells, whereby FGFR inhibition reduced the in vitro migratory capacity of N-cadherin-expressing cells, but not N-cadherin-negative cells [45, 84]. In addition, FGF-2 increased the invasiveness of N-cadherin-expressing MCF-7 human breast cancer cells, but not control MCF-7 cells [88]. To this end, it has been shown that N-cadherin potentiates FGF-2-activated FGFR-1 signalling by attenuating ligand-induced FGFR-1 internalisation, thereby stabilising FGFR-1 expression [111, 113]. In turn, the sustained activation of down-stream MEK/ERK signalling results in increased production of the extracellular matrix (ECM)-degrading enzyme matrix metalloproteinase-9 (MMP-9) and enhanced breast cancer cell invasiveness [88, 111]. In addition, the interaction of N-cadherin and FGFR is also likely to promote metastasis by activation of the phosphatidylinositide-3 kinase/Akt (PI3K/Akt) signalling pathway in some cancer cell types. For example, studies suggest that the invasiveness of N-cadherin-expressing ErbB2/Neu breast cancer cells following FGFR activation is mediated by PI3K/Akt signalling. N-cadherin potentiates FGFR-Akt signalling and sensitivity to FGFR inhibition in ErbB2/Neu cells, suggesting the involvement of an N-cadherin-FGFR-PI3K/Akt signalling axis in breast cancer cell invasion [115] (Fig. 2b).
Two lines of evidence suggest that N-cadherin-FGFR-1 interactions promote the invasive behaviour in both collectively migrating and individual cancer cells. Firstly, N-cadherin-FGFR-1 interactions have been shown to occur over most of the cell membrane, but are excluded from sites of cell-cell adhesion, suggesting that the interaction is independent of N-cadherin-mediated cellular adhesion [112]. Secondly, blocking antibodies directed at the FGFR-1-interacting domain of N-cadherin (EC4) have been shown to inhibit N-cadherin-mediated migration, but not N-cadherin-mediated aggregation, of human breast cancer cells [116]. Thus, it would appear that N-cadherin-mediated cell-cell adhesion and N-cadherin-mediated cell migration via FGFR-1 are independent and mutually exclusive events. Further studies are warranted to identify whether N-cadherin potentiates FGFR-1 signalling in other epithelial malignancies such as pancreatic cancer.
N-cadherin modulates canonical Wnt signalling
In addition to stabilising cadherin-mediated cell-cell adhesion, β-catenin plays a central role in the canonical Wnt signalling pathway. Canonical Wnt signalling promotes the cytoplasmic accumulation and nuclear translocation of β-catenin, which activates T cell factor/lymphoid enhancer factor (TCF/LEF)-mediated transcription of genes [117,118,119] that encode tumour invasion and metastasis-promoting molecules (e.g. MMPs and CD44) [120,121,122,123,124,125,126]. It has been proposed that cadherins and the canonical Wnt signalling pathway may compete for the same cellular pool of β-catenin, with cadherins sequestering β-catenin from the nucleus, thereby attenuating Wnt signalling [127, 128]. Indeed, enforced expression of N-cadherin in colon carcinoma cells resulted in the relocation of nuclear β-catenin to the plasma membrane and attenuated LEF-responsive trans-activation [129]. Alternatively, studies suggest that the N-cadherin-β-catenin complex may provide a stable pool of β-catenin available for TCF/LEF-mediated gene transcription in cancer cells [91, 130]. To this end, disruption of N-cadherin-mediated adhesion in leukaemic cells was found to increase TCF/LEF reporter activity [131]. Thus, given β-catenin is essential in the stabilisation of N-cadherin-mediated cellular adhesion (discussed earlier), it is feasible that the ability of N-cadherin to modulate TCF/LEF-mediated gene transcription may play an important role in individual cell migration, at the expense of collective cell migration (Fig. 2c).
Trans-endothelial migration is an important process in the haematogenous dissemination of cancer cells to distant sites [132]. Notably, studies suggest that N-cadherin promotes the trans-endothelial migration of cancer cells. To this end, N-cadherin silencing has been shown to reduce the ability of melanoma cells to undergo trans-endothelial migration in vitro [91]. Studies have demonstrated that N-cadherin-mediated melanoma cell adhesion to endothelial cells promotes trans-endothelial migration by modulating canonical Wnt signalling. β-catenin co-localises with N-cadherin during the initial stages of melanoma cell adhesion to endothelial cells; however, during transendothelial migration, the tyrosine kinase Src is activated and subsequently phosphorylates the N-cadherin cytoplasmic domain, thereby dissociating the N-cadherin-β-catenin complex. β-catenin is then translocated to the nucleus of melanoma cells and activates TCF/LEF-mediated gene transcription, resulting in up-regulation of the adhesion molecule CD44 [91, 133]. Studies using epithelial cancer cells suggest that CD44 binding to E-selectin on endothelial cells activates intracellular signalling pathways that lead to disassembly of endothelial junctions, thereby facilitating trans-endothelial migration [134,135,136]. In line with these studies, CD44 expression in melanoma cells has been shown to promote endothelial gap formation and trans-endothelial migration in vitro [137]. Moreover, N-cadherin knock-down in human melanoma cells reduces extravasation and lung nodule formation following intravenous injection in immuno-compromised mice [92]. Notably, while N-cadherin-expressing tumour cells have been detected in the circulation of patients with various epithelial cancers [59, 68, 76], and CD44 has been shown to promote diapedesis in breast cancer cells [134, 138], a role for N-cadherin in the trans-endothelial migration of epithelial cancer cells has not been directly demonstrated to date.
The emerging role of N-cadherin in haematological malignancies
We have thus far summarised the functional role and clinical implications of aberrant N-cadherin expression in the context of solid tumour metastasis. There is now emerging evidence suggesting that N-cadherin plays a role in haematological malignancies, including leukaemia and multiple myeloma (MM). These cancers account for approximately 10% of all cancer cases and are typically characterised by the abnormal proliferation of malignant white blood cells within the bone marrow (BM) and the presence of tumour cells within the circulation. Specialised compartments, or ‘niches’, within the BM microenvironment play critical roles in housing and maintaining pools of quiescent haematopoietic stem cells (HSCs), and in regulating HSC self-renewal and differentiation [139, 140]. Notably, N-cadherin is expressed by various cell types associated with the HSC niche, including osteoblasts and stromal cells in the endosteal niche, and endothelial cells and pericytes in the perivascular niche [32, 36, 141, 142]. In the following section, we discuss the potential implications of aberrant N-cadherin expression in haematological cancer cells; namely, BM homing and BM microenvironment-mediated protection to chemotherapeutic agents.
Leukaemia
Leukaemias are thought to arise by the malignant transformation of HSCs into leukaemic stem cells (LSCs) which occupy and modify BM HSC niches [143,144,145,146]. Adhesive interactions between LSCs and the BM microenvironment activate signalling cascades which contribute to LSC self-renewal and survival, and the capacity to evade the cytotoxic effects of chemotherapeutic agents [147, 148]. Indeed, therapeutic targeting of adhesion molecules to disrupt interactions with the niche represents a potential strategy to eliminate LSCs [149].
Studies have demonstrated that N-cadherin is expressed in a subpopulation of primitive HSCs [36], but its precise role within the HSC niche in normal haematopoiesis is controversial. To this end, the over-expression of N-cadherin in HSCs has been shown to increase HSC lodgement to BM endosteal surfaces in irradiated mice, enhance HSC self-renewal following serial BM transplantation and promote HSC quiescence in vitro [150]. However, other studies have reported that deletion of N-cadherin in HSCs or osteoblastic cells has no effect on haematopoiesis or HSC quiescence, self-renewal or long-term repopulating activity [141, 151, 152].
While these studies suggest that N-cadherin function may be dispensable in HSC niche maintenance, emerging evidence implicates N-cadherin in the function of the LSC niche. Studies have reported that N-cadherin is expressed on primitive sub-populations of leukaemic cells including patient-derived CD34+ CD38− chronic myeloid leukaemia (CML) cells and CD34+ CD38− CD123+ acute myeloid leukaemia (AML) cells, suggesting that N-cadherin is a marker of LSCs [130, 153, 154]. Similar to solid tumours, N-cadherin is thought to facilitate engagement of leukaemic cancer cells with cells of the surrounding BM microenvironment. For example, treatment of primary human CD34+ CML cells with the N-cadherin blocking antibody GC-4 significantly reduced their adhesion to human BM stromal cells (BMSCs) [130]. Similarly, GC-4 treatment of a BCR-ABL-positive mouse acute lymphoblastic leukaemia (ALL) cell line was found to inhibit their ability to adhere to mouse fibroblasts [155]. Pre-clinical mouse models also suggest that N-cadherin may promote BM homing, engraftment and self-renewal of AML cells in vivo [156, 157]. Thus, N-cadherin represents a potential target to inhibit LSC interactions with the BM microenvironment.
N-cadherin-mediated cell adhesive interactions promote microenvironmental protection of leukaemic cells to anti-cancer agents
Adhesive interactions between leukaemic cells and BMSCs confer sub-populations of leukaemic cells with resistance to anti-cancer agents, leading to disease relapse [158, 159]. As such, there is growing interest in targeting molecules involved in leukaemic cell-BMSC interactions to enhance leukaemic sensitivity to anti-cancer agents [130, 160]. The role of N-cadherin in the microenvironmental protection of leukaemic cells to anti-cancer agents was first demonstrated in studies showing that N-cadherin expression was associated with resistance to treatment with a farnesyltransferase inhibitor in the murine lymphoblastic leukaemia cell line, B-1, when grown in co-culture with fibroblasts. Enforced N-cadherin expression in B-1 cells also conferred farnesyltransferase inhibitor-resistance when grown in the presence of fibroblasts [155]. Notably, these findings are in line with reports showing that N-cadherin is up-regulated in solid tumour cancer cells resistant to anti-cancer agents [161,162,163,164] and androgen deprivation therapy [51, 165]. Direct demonstration that N-cadherin-mediated cell-cell adhesion facilitated microenvironmental protection of leukaemic cells to anti-cancer agents was provided in co-culture experiments with primary human CD34+ CML cells and BMSCs. Disruption of CML cell-BMSC adhesion, using an N-cadherin antagonist peptide (containing the HAV sequence) or the N-cadherin function-blocking antibody GC-4 increased CML cell sensitivity to the tyrosine kinase inhibitor imatinib [130, 131]. An association between response to chemotherapy and LSC expression of N-cadherin has also been reported in AML patients. To this end, studies suggest that AML patients exhibiting a higher proportion of N-cadherin-expressing BM-derived CD34+ CD38− CD123+ LSCs at diagnosis are less responsive to induction chemotherapy [153]. While the precise mechanism by which N-cadherin-mediated adhesion confers drug-resistance in leukaemic cells is unclear, studies in solid tumour cells suggest that N-cadherin-mediated adhesion increases activity of the anti-apoptotic protein Bcl-2, by PI3K/Akt-mediated inactivation of the pro-apoptotic protein Bad [86, 162, 166].
MM
MM is characterised by the uncontrolled proliferation of transformed immunoglobulin-producing plasma cells (PCs) within the BM. Data from our group, and others, suggest that N-cadherin gene and protein expression is elevated in CD138+ BM-derived PCs in approximately 50% of newly-diagnosed MM patients compared with BM PCs from healthy individuals and is associated with poor prognosis [81, 167] (Table 1). Notably, the expression of the N-cadherin gene, CDH2, is up-regulated in MM patients harbouring the high-risk t(4;14)(p16;q32) translocation [167, 168]. This translocation encompasses 15–20% of all MM patients and is universally characterised by the dysregulated expression of the oncogenic histone methyltransferase MMSET (also known as NSD2) [169,170,171]. In addition, CDH2 expression is also up-regulated in more than 50% of MM patients in the hyperdiploidy-related sub-group [167].
N-cadherin promotes MM PC BM homing
The progression of MM disease is underscored by MM PC egress from the primary BM environment and dissemination via the peripheral circulation to distal medullary sites [172]. Functionally, N-cadherin is thought to play a role in MM PC extravasation and homing to the BM. Following intravenous inoculation, the BM-homing capacity of the human MM PC line NCI-H929 in immuno-deficient mice was significantly attenuated by N-cadherin silencing in tumour cells, resulting in increased numbers of residual circulating tumour cells [167]. In addition, N-cadherin knock-down in the murine MM cell line 5TGM1 significantly inhibited adhesion to BM endothelial cell monolayers in vitro, although N-cadherin knock-down or GC-4 antibody-mediated blocking of N-cadherin did not affect the trans-endothelial migration capacity of MM PCs in vitro [167, 173]. Taken together, these data suggest that N-cadherin may promote BM homing of circulating MM PC by facilitating their adhesion to the vasculature, without affecting the rate of subsequent diapedesis.
N-cadherin mediates cell-cell adhesion between MM PCs and the BM microenvironment
Adhesive interactions between MM PCs and the BM microenvironment are critical in the permissiveness of the BM to the development of MM disease. These include cell-cell interactions which support MM PC growth and resistance to anti-cancer agents, and promote the inhibition of osteoblast differentiation, thereby contributing to MM PC-mediated bone loss [174, 175]. In addition to endothelial cell adhesion, in vitro studies have demonstrated that N-cadherin mediates the adhesion of human MM PCs to osteoblasts and stromal cells, which constitute the endosteal MM niche [167, 176]. In a functional context, N-cadherin-mediated adhesion between MM PCs and pre-osteoblastic cells has been shown to inhibit osteoblast differentiation, suggesting that N-cadherin may contribute to MM-related bone loss in the clinical setting [167]. Studies have also shown that treatment of human MM PC lines in co-culture with stromal cells or osteoblasts with the N-cadherin blocking antibody GC-4 induced a significant expansion of MM PCs in vitro [176]. Thus, it has been proposed N-cadherin may maintain the proliferative quiescence of MM PC in contact with cells of the endosteal MM niche [176]. In light of the role of N-cadherin in mediating leukaemic cell resistance to anti-cancer agents [130, 131, 155], these findings may provide a rationale to investigate whether N-cadherin-mediated adhesion potentiates resistance to anti-cancer agents in MM.
N-cadherin as a therapeutic target in cancer
As N-cadherin is widely implicated in cancer metastasis, the utility of N-cadherin antagonists as therapeutic drugs is being investigated in the oncology setting. Notably, N-cadherin-targeting agents have been shown to inhibit cell adhesion and to modulate cell signalling. Interestingly, studies have also shown that N-cadherin-targeting agents affect both tumour cells and tumour-associated vasculature. Here, we describe the current repertoire of N-cadherin antagonists that have displayed efficacy as anti-cancer agents in vivo.
Monoclonal antibodies
Several monoclonal antibodies directed against N-cadherin have been investigated for their ability to block N-cadherin-dependent tumour migration and invasion in vitro and metastasis in vivo. The mouse monoclonal antibody, designated GC-4, binds to the EC1 domain of N-cadherin monomers and subsequently blocks N-cadherin-mediated adhesion [36, 167, 177, 178]. GC-4 has been shown to suppress N-cadherin-mediated Akt signalling [61, 166], and inhibit the migration and invasion of melanoma, bladder, ovarian and breast cancer cells in vitro [61, 87, 88, 91]. In addition, pre-treatment of AML cells with GC-4 has been shown to inhibit BM homing of circulating tumour cells in vivo [156]. Thus, as N-cadherin plays a role in trans-endothelial migration and BM homing of circulating tumour cells in melanoma and MM, in addition to AML [91, 156, 167, 173], treatment with GC-4 may by therapeutically relevant in the context of limiting the metastatic dissemination of tumour cells in these cancers. Additionally, GC-4-mediated blocking of N-cadherin engagement between human CD34+ CML cells and stromal cells increased tumour cell sensitivity to imatinib, demonstrating a potential therapeutic strategy to overcome tyrosine kinase inhibitor resistance [131]. Two additional monoclonal antibodies, 1H7 (targeting N-cadherin EC1–3) and 2A9 (targeting N-cadherin EC4), have shown efficacy in a subcutaneous xenograft prostate cancer mouse model, whereby both antibodies reduced the growth of established tumours and inhibited localised muscle invasion and distant lymph node metastasis [89].
ADH-1
The lateral clustering of N-cadherin monomers (cis adhesion) is essential in the stabilisation and maturation of nascent N-cadherin-mediated adhesive junctions between neighbouring cells [14, 16]. Peptides containing the classical cadherin motif, HAV, are likely to compete with the HAV motif on N-cadherin EC1 for binding to a recognition sequence on EC2 of an adjacent N-cadherin monomer, thereby inhibiting the lateral clustering of N-cadherin monomers [179]. On the basis that a HAV motif located on FGFR-1 is required for FGF-2 binding [112], it is feasible that peptides containing a HAV motif may also inhibit FGFR signalling. This concept led to the development of ADH-1 (N-Ac-CHAVC-NH2), a stable cyclic peptide harbouring a HAV motif, which similarly inhibited N-cadherin-dependent function [180]. In vitro, ADH-1 has been shown to induce apoptosis in a range of tumour cell types, and inhibits tumour cell migration at sub-cytotoxic concentrations, with cell sensitivity proportional to relative N-cadherin expression [181,182,183]. The efficacy of ADH-1 as an anti-cancer agent has been demonstrated in a number of pre-clinical mouse models including pancreatic, breast, colon, ovarian and lung cancer [181, 184]. In addition to inhibiting primary tumour growth, pre-clinical studies also suggest that ADH-1 may inhibit localised tumour invasion and dissemination via the circulation [173, 181]. For example, studies using a mouse model of MM reported that daily ADH-1 treatment commencing immediately prior to, but not after, intravenous inoculation of MM PCs resulted in inhibition of tumour development [173]. Notably, ADH-1 has also been identified as a vascular-disrupting agent, suggesting the compound may have effects on both tumour cells and tumour-associated vasculature [184, 185]. In phase I clinical trials, ADH-1 was shown to have an acceptable toxicity profile with no maximum tolerated dose achieved. ADH-1 treatment was associated with disease control in approximately 25% of patients with advanced chemotherapy-refractory solid tumours, independent of tumour N-cadherin expression status [186, 187].
The therapeutic efficacy of ADH-1 as an anti-cancer agent has been most extensively evaluated in the melanoma setting. Pre-clinical studies suggest that ADH-1 synergistically enhances melanoma tumour response to melphalan [188, 189]. These studies showed that ADH-1 enhances the permeability of tumour vasculature and increases melphalan delivery to the tumour microenvironment, as evidenced by increased formation of melphalan-DNA adducts in tumours. However, the combinatorial effects of ADH-1 and melphalan were not replicated in phase I/II clinical trials [190, 191]. In contrast to other tumour settings, studies have also suggested that ADH-1 may stimulate tumour growth in some mouse models of melanoma [188, 189]. These effects were associated with activation of pro-growth and survival intracellular signalling pathways including Akt signalling and the down-stream mTOR signalling pathway in vitro and in vivo [189]. These data suggest that ADH-1 may act as an N-cadherin agonist in certain tumour contexts. However, to date, ADH-1-mediated activation of tumour cell proliferation and signalling has not been reported in the clinical setting.
Conclusions
The up-regulation or ‘de novo’ expression of N-cadherin has significant negative implications in metastasis-related cancer relapse and progression, as well as overall survival of cancer patients. In addition to its prognostic significance in cancer, N-cadherin actively promotes the metastatic capacity of tumour cells. Here, we have described three distinct mechanisms by which N-cadherin endows tumour cells with increased migratory capacity: facilitation of collective cell migration, augmentation of FGFR-1 signalling and modulation of canonical Wnt signalling. Unfortunately, our understanding of how N-cadherin influences cancer cell metastasis, and tumorigenesis in general, remains incomplete. Studies in cardiomyocytes, stromal cells and epithelial cancer-like cells have ascribed focal adhesion-like properties to N-cadherin including mechanotransduction and traction-force transmission [192,193,194,195]. Indeed, whether a ‘traction and propulsion’-type system, via homotypic N-cadherin mediated cell-cell contacts, is utilised by cancer cells to facilitate migration is intriguing and warrants further investigation. Moreover, there is an emerging body of evidence demonstrating that N-cadherin is expressed and is functionally relevant in the context of numerous haematological malignancies including lymphoblastic and myelogenous leukaemias, and MM. Functionally, pre-clinical studies have demonstrated that N-cadherin promotes the BM homing capacity of circulating MM and leukaemic cells, thereby facilitating metastatic dissemination and intramedullary tumour colonisation [156, 167, 173]. Given N-cadherin is expressed by circulating tumour cells in several epithelial cancers [59, 68, 76] and facilitates trans-endothelial migration in melanoma cells [91, 133], it is tempting to speculate that N-cadherin may also promote tumour cell extravasation in non-haematological malignancies. Studies also suggest that N-cadherin facilitates engagement of LSCs with the tumour microenvironment and promotes leukaemic cell resistance to anti-cancer agents [130, 131, 155]. On the basis of observations in epithelial cancers, N-cadherin may mediate drug resistance in leukaemic cells, at least in part, by activation of the pro-survival protein Bcl-2 [89, 162, 166], or modulation of Sonic Hedgehog signalling [196], widely implicated in cancer stem cell function and maintenance [197]. Interestingly, N-cadherin expression is induced in solid tumour cells resistant to standard anti-cancer agents including tyrosine kinase inhibitors [161,162,163,164]. However, it remains to be determined whether N-cadherin functionally contributes to microenvironmental cell adhesion mediated-drug resistance in these cancers.
Given the established role of N-cadherin in cancer, N-cadherin is continually being investigated as a therapeutic target. To date, peptides and mouse monoclonal antibodies have demonstrated some efficacy in the pre-clinical setting, by inhibiting cancer metastasis, enhancing cancer cell sensitivity to chemotherapeutic agents and delaying castration resistance in prostate cancer. However, the challenge remains to develop N-cadherin antagonists which are effective anti-cancer agents in the clinical setting. The humanisation of N-cadherin-blocking antibodies such as GC-4 may represent one such approach to utilise N-cadherin as a therapeutic target. Moreover, the development of next-generation N-cadherin-targeting small molecules with enhanced stability over existing peptide inhibitors show promise as potent inhibitors of N-cadherin function [198,199,200]. It remains to be seen whether these compounds have efficacy as anti-cancer agents. Undoubtedly, further exploration of N-cadherin as a therapeutic target to inhibit metastasis and overcome drug resistance is warranted.
Abbreviations
- ALL:
-
Acute lymphoblastic leukaemia
- AML:
-
Acute myeloid leukaemia
- BM:
-
Bone marrow
- BMSC:
-
Bone marrow stromal cell
- CML:
-
Chronic myeloid leukaemia
- EC1:
-
First extracellular domain
- EC2:
-
Second extracellular domain
- EC4:
-
Fourth extracellular domain
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial-to-mesenchymal transition
- FGFR:
-
Fibroblast growth factor-receptor
- HAV:
-
Histidine-alanine-valine
- HSC:
-
Haematopoietic stem cell
- LSC:
-
Leukaemic stem cells
- MDCK:
-
Transformed canine kidney epithelial cells
- MM:
-
Multiple myeloma
- PC:
-
Plasma cell
- PI3K:
-
Phosphatidylinositide-3 kinase
- TCF/LEF:
-
T cell factor/lymphoid enhancer factor
References
Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–92.
Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147(5):992–1009.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90.
De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110.
Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR. Cadherin switching. J Cell Sci. 2008;121(Pt 6):727–35.
Gheldof A, Berx G. Cadherins and epithelial-to-mesenchymal transition. Prog Mol Biol Transl Sci. 2013;116:317–36.
Lade-Keller J, Riber-Hansen R, Guldberg P, Schmidt H, Hamilton-Dutoit SJ, Steiniche T. E- to N-cadherin switch in melanoma is associated with decreased expression of phosphatase and tensin homolog and cancer progression. Br J Dermatol. 2013;169(3):618–28.
Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res. 2007;13(23):7003–11.
Araki K, Shimura T, Suzuki H, Tsutsumi S, Wada W, Yajima T, Kobayahi T, Kubo N, Kuwano H. E/N-cadherin switch mediates cancer progression via TGF-beta-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br J Cancer. 2011;105(12):1885–93.
Aleskandarany MA, Negm OH, Green AR, Ahmed MA, Nolan CC, Tighe PJ, Ellis IO, Rakha EA. Epithelial mesenchymal transition in early invasive breast cancer: an immunohistochemical and reverse phase protein array study. Breast Cancer Res Treat. 2014;145(2):339–48.
Kourtidis A, Lu R, Pence LJ, Anastasiadis PZ. A central role for cadherin signaling in cancer. Exp Cell Res. 2017;358(1):78–85.
Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392(6672):190–3.
Hazan RB, Qiao R, Keren R, Badano I, Suyama K. Cadherin switch in tumor progression. Ann N Y Acad Sci. 2004;1014:155–63.
Harrison OJ, Jin X, Hong S, Bahna F, Ahlsen G, Brasch J, Wu Y, Vendome J, Felsovalyi K, Hampton CM, et al. The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure. 2011;19(2):244–56.
Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, Legrand JF, Als-Nielsen J, Colman DR, Hendrickson WA. Structural basis of cell-cell adhesion by cadherins. Nature. 1995;374(6520):327–37.
Yap AS, Brieher WM, Pruschy M, Gumbiner BM. Lateral clustering of the adhesive ectodomain: a fundamental determinant of cadherin function. Curr Biol. 1997;7(5):308–15.
Taulet N, Comunale F, Favard C, Charrasse S, Bodin S, Gauthier-Rouviere C. N-cadherin/p120 catenin association at cell-cell contacts occurs in cholesterol-rich membrane domains and is required for RhoA activation and myogenesis. J Biol Chem. 2009;284(34):23137–45.
Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol. 2003;163(3):525–34.
Yap AS, Kovacs EM. Direct cadherin-activated cell signaling: a view from the plasma membrane. J Cell Biol. 2003;160(1):11–6.
Ratheesh A, Priya R, Yap AS. Coordinating rho and Rac: the regulation of Rho GTPase signaling and cadherin junctions. Prog Mol Biol Transl Sci. 2013;116:49–68.
Charrasse S, Meriane M, Comunale F, Blangy A, Gauthier-Rouviere C. N-cadherin-dependent cell-cell contact regulates rho GTPases and beta-catenin localization in mouse C2C12 myoblasts. J Cell Biol. 2002;158(5):953–65.
Comunale F, Causeret M, Favard C, Cau J, Taulet N, Charrasse S, Gauthier-Rouviere C. Rac1 and RhoA GTPases have antagonistic functions during N-cadherin-dependent cell-cell contact formation in C2C12 myoblasts. Biol Cell. 2007;99(9):503–17.
Niessen CM, Leckband D, Yap AS. Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol Rev. 2011;91(2):691–731.
Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu Rev Cell Dev Biol. 2007;23:237–61.
McLachlan RW, Yap AS. Not so simple: the complexity of phosphotyrosine signaling at cadherin adhesive contacts. J Mol Med (Berl). 2007;85(6):545–54.
Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol. 2005;17(5):459–65.
Guo HB, Johnson H, Randolph M, Pierce M. Regulation of homotypic cell-cell adhesion by branched N-glycosylation of N-cadherin extracellular EC2 and EC3 domains. J Biol Chem. 2009;284(50):34986–97.
Takeichi M. The cadherins: cell-cell adhesion molecules controlling animal morphogenesis. Development. 1988;102(4):639–55.
Radice GL, Rayburn H, Matsunami H, Knudsen KA, Takeichi M, Hynes RO. Developmental defects in mouse embryos lacking N-cadherin. Dev Biol. 1997;181(1):64–78.
Fontana F, Hickman-Brecks CL, Salazar VS, Revollo L, Abou-Ezzi G, Grimston SK, Jeong SY, Watkins M, Fortunato M, Alippe Y, et al. N-cadherin regulation of bone growth and homeostasis is Osteolineage stage-specific. J Bone Miner Res. 2017;32(6):1332–42.
George-Weinstein M, Gerhart J, Blitz J, Simak E, Knudsen KA. N-cadherin promotes the commitment and differentiation of skeletal muscle precursor cells. Dev Biol. 1997;185(1):14–24.
Paik JH, Skoura A, Chae SS, Cowan AE, Han DK, Proia RL, Hla T. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev. 2004;18(19):2392–403.
Williams EJ, Furness J, Walsh FS, Doherty P. Activation of the FGF receptor underlies neurite outgrowth stimulated by L1, N-CAM, and N-cadherin. Neuron. 1994;13(3):583–94.
Tanaka H, Shan W, Phillips GR, Arndt K, Bozdagi O, Shapiro L, Huntley GW, Benson DL, Colman DR. Molecular modification of N-cadherin in response to synaptic activity. Neuron. 2000;25(1):93–107.
Alimperti S, Mirabella T, Bajaj V, Polacheck W, Pirone DM, Duffield J, Eyckmans J, Assoian RK, Chen CS. Three-dimensional biomimetic vascular model reveals a RhoA, Rac1, and N-cadherin balance in mural cell-endothelial cell-regulated barrier function. Proc Natl Acad Sci U S A. 2017;114(33):8758–63.
Puch S, Armeanu S, Kibler C, Johnson KR, Muller CA, Wheelock MJ, Klein G. N-cadherin is developmentally regulated and functionally involved in early hematopoietic cell differentiation. J Cell Sci. 2001;114(Pt 8):1567–77.
Tomita K, van Bokhoven A, van Leenders GJ, Ruijter ET, Jansen CF, Bussemakers MJ, Schalken JA. Cadherin switching in human prostate cancer progression. Cancer Res. 2000;60(13):3650–4.
Choi Y, Lee HJ, Jang MH, Gwak JM, Lee KS, Kim EJ, Kim HJ, Lee HE, Park SY. Epithelial-mesenchymal transition increases during the progression of in situ to invasive basal-like breast cancer. Hum Pathol. 2013;44(11):2581–9.
Lascombe I, Clairotte A, Fauconnet S, Bernardini S, Wallerand H, Kantelip B, Bittard H. N-cadherin as a novel prognostic marker of progression in superficial urothelial tumors. Clin Cancer Res. 2006;12(9):2780–7.
Nakajima S, Doi R, Toyoda E, Tsuji S, Wada M, Koizumi M, Tulachan SS, Ito D, Kami K, Mori T, et al. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin Cancer Res. 2004;10(12 Pt 1):4125–33.
Hsu MY, Wheelock MJ, Johnson KR, Herlyn M. Shifts in cadherin profiles between human normal melanocytes and melanomas. J Investig Dermatol Symp Proc. 1996;1(2):188–94.
Hao L, Ha JR, Kuzel P, Garcia E, Persad S. Cadherin switch from E- to N-cadherin in melanoma progression is regulated by the PI3K/PTEN pathway through twist and snail. Br J Dermatol. 2012;166(6):1184–97.
Watson-Hurst K, Becker D. The role of N-cadherin, MCAM and beta3 integrin in melanoma progression, proliferation, migration and invasion. Cancer Biol Ther. 2006;5(10):1375–82.
Knudsen KA, Sauer C, Johnson KR, Wheelock MJ. Effect of N-cadherin misexpression by the mammary epithelium in mice. J Cell Biochem. 2005;95(6):1093–107.
Hulit J, Suyama K, Chung S, Keren R, Agiostratidou G, Shan W, Dong X, Williams TM, Lisanti MP, Knudsen K, et al. N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res. 2007;67(7):3106–16.
Su Y, Li J, Shi C, Hruban RH, Radice GL. N-cadherin functions as a growth suppressor in a model of K-ras-induced PanIN. Oncogene. 2016;35(25):3335–41.
Saadatmand S, de Kruijf EM, Sajet A, Dekker-Ensink NG, van Nes JG, Putter H, Smit VT, van de Velde CJ, Liefers GJ, Kuppen PJ. Expression of cell adhesion molecules and prognosis in breast cancer. Br J Surg. 2013;100(2):252–60.
Aleskandarany MA, Soria D, Green AR, Nolan C, Diez-Rodriguez M, Ellis IO, Rakha EA. Markers of progression in early-stage invasive breast cancer: a predictive immunohistochemical panel algorithm for distant recurrence risk stratification. Breast Cancer Res Treat. 2015;151(2):325–33.
Bock C, Kuhn C, Ditsch N, Krebold R, Heublein S, Mayr D, Doisneau-Sixou S, Jeschke U. Strong correlation between N-cadherin and CD133 in breast cancer: role of both markers in metastatic events. J Cancer Res Clin Oncol. 2014;140(11):1873–81.
Ning Q, Liu C, Hou L, Meng M, Zhang X, Luo M, Shao S, Zuo X, Zhao X. Vascular endothelial growth factor receptor-1 activation promotes migration and invasion of breast cancer cells through epithelial-mesenchymal transition. PLoS One. 2013;8(6):e65217.
Jennbacken K, Tesan T, Wang W, Gustavsson H, Damber JE, Welen K. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr Relat Cancer. 2010;17(2):469–79.
Drivalos A, Chrisofos M, Efstathiou E, Kapranou A, Kollaitis G, Koutlis G, Antoniou N, Karanastasis D, Dimopoulos MA, Bamias A. Expression of alpha5-integrin, alpha7-integrin, Epsilon-cadherin, and N-cadherin in localized prostate cancer. Urol Oncol. 2016;34(4):165.e111–68.
Derycke L, De Wever O, Stove V, Vanhoecke B, Delanghe J, Depypere H, Bracke M. Soluble N-cadherin in human biological fluids. Int J Cancer. 2006;119(12):2895–900.
Ge R, Wang Z, Wu S, Zhuo Y, Otsetov AG, Cai C, Zhong W, Wu CL, Olumi AF. Metformin represses cancer cells via alternate pathways in N-cadherin expressing vs. N-cadherin deficient cells. Oncotarget. 2015;6(30):28973–87.
Hui L, Zhang S, Dong X, Tian D, Cui Z, Qiu X. Prognostic significance of twist and N-cadherin expression in NSCLC. PLoS One. 2013;8(4):e62171.
Xu J, Lv W, Hu Y, Wang L, Wang Y, Cao J, Hu J. Wnt3a expression is associated with epithelial-mesenchymal transition and impacts prognosis of lung adenocarcinoma patients. J Cancer. 2017;8(13):2523–31.
Mo D, Yang D, Xiao X, Sun R, Huang L, Xu J. MiRNA-145 suppresses lung adenocarcinoma cell invasion and migration by targeting N-cadherin. Biotechnol Lett. 2017;39(5):701–10.
Yang Z, Wang H, Xia L, Oyang L, Zhou Y, Zhang B, Chen X, Luo X, Liao Q, Liang J. Overexpression of PAK1 correlates with aberrant expression of EMT markers and poor prognosis in non-small cell lung Cancer. J Cancer. 2017;8(8):1484–91.
Nel I, Jehn U, Gauler T, Hoffmann AC. Individual profiling of circulating tumor cell composition in patients with non-small cell lung cancer receiving platinum based treatment. Transl Lung Cancer Res. 2014;3(2):100–6.
Abufaraj M, Haitel A, Moschini M, Gust K, Foerster B, Ozsoy M, D'Andrea D, Karakiewicz PI, Roupret M, Briganti A, et al. Prognostic role of N-cadherin expression in patients with invasive bladder Cancer. Clin Genitourin Cancer. 2017. https://doi.org/10.1016/j.clgc.2017.07.001.
Wallerand H, Cai Y, Wainberg ZA, Garraway I, Lascombe I, Nicolle G, Thiery JP, Bittard H, Radvanyi F, Reiter RR. Phospho-Akt pathway activation and inhibition depends on N-cadherin or phospho-EGFR expression in invasive human bladder cancer cell lines. Urol Oncol. 2010;28(2):180–8.
Muramaki M, Miyake H, Terakawa T, Kumano M, Sakai I, Fujisawa M. Expression profile of E-cadherin and N-cadherin in non-muscle-invasive bladder cancer as a novel predictor of intravesical recurrence following transurethral resection. Urol Oncol. 2012;30(2):161–6.
Muramaki M, Miyake H, Terakawa T, Kusuda Y, Fujisawa M. Expression profile of E-cadherin and N-cadherin in urothelial carcinoma of the upper urinary tract is associated with disease recurrence in patients undergoing nephroureterectomy. Urology. 2011;78(6):1443.e1447–12.
Zhou SJ, Liu FY, Zhang AH, Liang HF, Wang Y, Ma R, Jiang YH, Sun NF. MicroRNA-199b-5p attenuates TGF-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Br J Cancer. 2017;117(2):233–44.
Seo DD, Lee HC, Kim HJ, Min HJ, Kim KM, Lim YS, Chung YH, Lee YS, Suh DJ, Yu E, et al. Neural cadherin overexpression is a predictive marker for early postoperative recurrence in hepatocellular carcinoma patients. J Gastroenterol Hepatol. 2008;23(7 Pt 1):1112–8.
Yao X, Wang X, Wang Z, Dai L, Zhang G, Yan Q, Zhou W. Clinicopathological and prognostic significance of epithelial mesenchymal transition-related protein expression in intrahepatic cholangiocarcinoma. Onco Targets Ther. 2012;5:255–61.
Zhang J, Cheng Q, Zhou Y, Wang Y, Chen X. Slug is a key mediator of hypoxia induced cadherin switch in HNSCC: correlations with poor prognosis. Oral Oncol. 2013;49(11):1043–50.
Weller P, Nel I, Hassenkamp P, Gauler T, Schlueter A, Lang S, Dountsop P, Hoffmann AC, Lehnerdt G. Detection of circulating tumor cell subpopulations in patients with head and neck squamous cell carcinoma (HNSCC). PLoS One. 2014;9(12):e113706.
Mezi S, Chiappetta C, Carletti R, Nardini A, Cortesi E, Orsi E, Piesco G, Di Gioia C. Clinical significance of epithelial-to-mesenchymal transition in laryngeal carcinoma: its role in the different subsites. Head Neck. 2017;39(9):1806–18.
Luo WR, Wu AB, Fang WY, Li SY, Yao KT. Nuclear expression of N-cadherin correlates with poor prognosis of nasopharyngeal carcinoma. Histopathology. 2012;61(2):237–46.
Ye Z, Zhou M, Tian B, Wu B, Li J. Expression of lncRNA-CCAT1, E-cadherin and N-cadherin in colorectal cancer and its clinical significance. Int J Clin Exp Med. 2015;8(3):3707–15.
Yan X, Yan L, Liu S, Shan Z, Tian Y, Jin Z. N-cadherin, a novel prognostic biomarker, drives malignant progression of colorectal cancer. Mol Med Rep. 2015;12(2):2999–3006.
Liu CC, Cai DL, Sun F, Wu ZH, Yue B, Zhao SL, Wu XS, Zhang M, Zhu XW, Peng ZH, et al. FERMT1 mediates epithelial-mesenchymal transition to promote colon cancer metastasis via modulation of beta-catenin transcriptional activity. Oncogene. 2017;36(13):1779–92.
Okubo K, Uenosono Y, Arigami T, Yanagita S, Matsushita D, Kijima T, Amatatsu M, Uchikado Y, Kijima Y, Maemura K, et al. Clinical significance of altering epithelial-mesenchymal transition in metastatic lymph nodes of gastric cancer. Gastric Cancer. 2017;20(5):802–10.
Kamikihara T, Ishigami S, Arigami T, Matsumoto M, Okumura H, Uchikado Y, Kita Y, Kurahara H, Kijima Y, Ueno S, et al. Clinical implications of N-cadherin expression in gastric cancer. Pathol Int. 2012;62(3):161–6.
Nel I, Gauler TC, Bublitz K, Lazaridis L, Goergens A, Giebel B, Schuler M, Hoffmann AC. Circulating tumor cell composition in renal cell carcinoma. PLoS One. 2016;11(4):e0153018.
Quattrocchi L, Green AR, Martin S, Durrant L, Deen S. The cadherin switch in ovarian high-grade serous carcinoma is associated with disease progression. Virchows Arch. 2011;459(1):21–9.
Yi S, Yang ZL, Miao X, Zou Q, Li J, Liang L, Zeng G, Chen S. N-cadherin and P-cadherin are biomarkers for invasion, metastasis, and poor prognosis of gallbladder carcinomas. Pathol Res Pract. 2014;210(6):363–8.
Han K, Zhao T, Chen X, Bian N, Yang T, Ma Q, Cai C, Fan Q, Zhou Y, Ma B. microRNA-194 suppresses osteosarcoma cell proliferation and metastasis in vitro and in vivo by targeting CDH2 and IGF1R. Int J Oncol. 2014;45(4):1437–49.
Niimi R, Matsumine A, Iino T, Nakazora S, Nakamura T, Uchida A, Sudo A. Soluble neural-cadherin as a novel biomarker for malignant bone and soft tissue tumors. BMC Cancer. 2013;13(1):309.
Vandyke K, Chow AW, Williams SA, To LB, Zannettino AC. Circulating N-cadherin levels are a negative prognostic indicator in patients with multiple myeloma. Br J Haematol. 2013;161(4):499–507.
Luo Y, Yu T, Zhang Q, Fu Q, Hu Y, Xiang M, Peng H, Zheng T, Lu L, Shi H. Up-regulated N-cadherin expression is associated with poor prognosis in epithelial-derived solid tumors: a meta-analysis. Eur J Clin Investig. 2018. https://doi.org/10.1111/eci.12903.
Islam S, Carey TE, Wolf GT, Wheelock MJ, Johnson KR. Expression of N-cadherin by human squamous carcinoma cells induces a scattered fibroblastic phenotype with disrupted cell-cell adhesion. J Cell Biol. 1996;135(6 Pt 1):1643–54.
Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol. 1999;147(3):631–44.
Shintani Y, Hollingsworth MA, Wheelock MJ, Johnson KR. Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH(2)-terminal kinase 1 and up-regulating N-cadherin expression. Cancer Res. 2006;66(24):11745–53.
Li G, Satyamoorthy K, Herlyn M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001;61(9):3819–25.
Klymenko Y, Kim O, Loughran E, Yang J, Lombard R, Alber M, Stack MS. Cadherin composition and multicellular aggregate invasion in organotypic models of epithelial ovarian cancer intraperitoneal metastasis. Oncogene. 2017;36(42):5840–51.
Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 2000;148(4):779–90.
Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, Fazli L, Wada R, Huang J, Vessella RL, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. 2010;16(12):1414–20.
Sandig M, Voura EB, Kalnins VI, Siu CH. Role of cadherins in the transendothelial migration of melanoma cells in culture. Cell Motil Cytoskeleton. 1997;38(4):351–64.
Qi J, Chen N, Wang J, Siu CH. Transendothelial migration of melanoma cells involves N-cadherin-mediated adhesion and activation of the beta-catenin signaling pathway. Mol Biol Cell. 2005;16(9):4386–97.
Na YR, Lee JS, Lee SJ, Seok SH. Interleukin-6-induced twist and N-cadherin enhance melanoma cell metastasis. Melanoma Res. 2013;23(6):434–43.
Etienne-Manneville S. Neighborly relations during collective migration. Curr Opin Cell Biol. 2014;30:51–9.
Barriga EH, Mayor R. Adjustable viscoelasticity allows for efficient collective cell migration. Semin Cell Dev Biol. 2018. https://doi.org/10.1016/j.semcdb.2018.05.027.
Clark AG, Vignjevic DM. Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol. 2015;36:13–22.
Mayor R, Etienne-Manneville S. The front and rear of collective cell migration. Nat Rev Mol Cell Biol. 2016;17(2):97–109.
De Pascalis C, Etienne-Manneville S. Single and collective cell migration: the mechanics of adhesions. Mol Biol Cell. 2017;28(14):1833–46.
Kuriyama S, Yoshida M, Yano S, Aiba N, Kohno T, Minamiya Y, Goto A, Tanaka M. LPP inhibits collective cell migration during lung cancer dissemination. Oncogene. 2016;35(8):952–64.
Shih W, Yamada S. N-cadherin-mediated cell-cell adhesion promotes cell migration in a three-dimensional matrix. J Cell Sci. 2012;125(Pt 15):3661–70.
Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 2015;36:103–12.
Combedazou A, Gayral S, Colombie N, Fougerat A, Laffargue M, Ramel D. Small GTPases orchestrate cell-cell communication during collective cell movement. Small GTPases. 2017. https://doi.org/10.1080/21541248.2017.1366965.
Sabatini PJ, Zhang M, Silverman-Gavrila R, Bendeck MP, Langille BL. Homotypic and endothelial cell adhesions via N-cadherin determine polarity and regulate migration of vascular smooth muscle cells. Circ Res. 2008;103(4):405–12.
Theveneau E, Marchant L, Kuriyama S, Gull M, Moepps B, Parsons M, Mayor R. Collective chemotaxis requires contact-dependent cell polarity. Dev Cell. 2010;19(1):39–53.
Ouyang M, Lu S, Kim T, Chen CE, Seong J, Leckband DE, Wang F, Reynolds AB, Schwartz MA, Wang Y. N-cadherin regulates spatially polarized signals through distinct p120ctn and beta-catenin-dependent signalling pathways. Nat Commun. 2013;4:1589.
Wasil LR, Shair KH. Epstein-Barr virus LMP1 induces focal adhesions and epithelial cell migration through effects on integrin-alpha5 and N-cadherin. Oncogenesis. 2015;4:e171.
Siret C, Terciolo C, Dobric A, Habib MC, Germain S, Bonnier R, Lombardo D, Rigot V, Andre F. Interplay between cadherins and alpha2beta1 integrin differentially regulates melanoma cell invasion. Br J Cancer. 2015;113(10):1445–53.
Yano H, Mazaki Y, Kurokawa K, Hanks SK, Matsuda M, Sabe H. Roles played by a subset of integrin signaling molecules in cadherin-based cell-cell adhesion. J Cell Biol. 2004;166(2):283–95.
Moreno-Layseca P, Streuli CH. Signalling pathways linking integrins with cell cycle progression. Matrix Biol. 2014;34:144–53.
Grande-Garcia A, Echarri A, Del Pozo MA. Integrin regulation of membrane domain trafficking and Rac targeting. Biochem Soc Trans. 2005;33(Pt 4):609–13.
Williams EJ, Williams G, Howell FV, Skaper SD, Walsh FS, Doherty P. Identification of an N-cadherin motif that can interact with the fibroblast growth factor receptor and is required for axonal growth. J Biol Chem. 2001;276(47):43879–86.
Suyama K, Shapiro I, Guttman M, Hazan RB. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell. 2002;2(4):301–14.
Sanchez-Heras E, Howell FV, Williams G, Doherty P. The fibroblast growth factor receptor acid box is essential for interactions with N-cadherin and all of the major isoforms of neural cell adhesion molecule. J Biol Chem. 2006;281(46):35208–16.
Rachagani S, Macha MA, Ponnusamy MP, Haridas D, Kaur S, Jain M, Batra SK. MUC4 potentiates invasion and metastasis of pancreatic cancer cells through stabilization of fibroblast growth factor receptor 1. Carcinogenesis. 2012;33(10):1953–64.
Cavallaro U, Niedermeyer J, Fuxa M, Christofori G. N-CAM modulates tumour-cell adhesion to matrix by inducing FGF-receptor signalling. Nat Cell Biol. 2001;3(7):650–7.
Qian X, Anzovino A, Kim S, Suyama K, Yao J, Hulit J, Agiostratidou G, Chandiramani N, McDaid HM, Nagi C, et al. N-cadherin/FGFR promotes metastasis through epithelial-to-mesenchymal transition and stem/progenitor cell-like properties. Oncogene. 2014;33(26):3411–21.
Kim JB, Islam S, Kim YJ, Prudoff RS, Sass KM, Wheelock MJ, Johnson KR. N-cadherin extracellular repeat 4 mediates epithelial to mesenchymal transition and increased motility. J Cell Biol. 2000;151(6):1193–206.
Murillo-Garzon V, Kypta R. WNT signalling in prostate cancer. Nat Rev Urol. 2017;14(11):683–96.
Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. EMBO J. 2012;31(12):2714–36.
Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8(5):387–98.
Qi J, Yu Y, Akilli Ozturk O, Holland JD, Besser D, Fritzmann J, Wulf-Goldenberg A, Eckert K, Fichtner I, Birchmeier W. New Wnt/beta-catenin target genes promote experimental metastasis and migration of colorectal cancer cells through different signals. Gut. 2016;65(10):1690–701.
Stein U, Arlt F, Walther W, Smith J, Waldman T, Harris ED, Mertins SD, Heizmann CW, Allard D, Birchmeier W, et al. The metastasis-associated gene S100A4 is a novel target of beta-catenin/T-cell factor signaling in colon cancer. Gastroenterology. 2006;131(5):1486–500.
Zeilstra J, Joosten SP, Dokter M, Verwiel E, Spaargaren M, Pals ST. Deletion of the WNT target and cancer stem cell marker CD44 in Apc(min/+) mice attenuates intestinal tumorigenesis. Cancer Res. 2008;68(10):3655–61.
Crawford HC, Fingleton BM, Rudolph-Owen LA, Goss KJ, Rubinfeld B, Polakis P, Matrisian LM. The metalloproteinase matrilysin is a target of beta-catenin transactivation in intestinal tumors. Oncogene. 1999;18(18):2883–91.
Chen L, Li M, Li Q, Wang CJ, Xie SQ. DKK1 promotes hepatocellular carcinoma cell migration and invasion through beta-catenin/MMP7 signaling pathway. Mol Cancer. 2013;12:157.
Lowy AM, Clements WM, Bishop J, Kong L, Bonney T, Sisco K, Aronow B, Fenoglio-Preiser C, Groden J. Beta-catenin/Wnt signaling regulates expression of the membrane type 3 matrix metalloproteinase in gastric cancer. Cancer Res. 2006;66(9):4734–41.
Takahashi M, Tsunoda T, Seiki M, Nakamura Y, Furukawa Y. Identification of membrane-type matrix metalloproteinase-1 as a target of the beta-catenin/Tcf4 complex in human colorectal cancers. Oncogene. 2002;21(38):5861–7.
Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol. 2010;2(2):a002915.
Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303(5663):1483–7.
Sadot E, Simcha I, Shtutman M, Ben-Ze'ev A, Geiger B. Inhibition of beta-catenin-mediated transactivation by cadherin derivatives. Proc Natl Acad Sci U S A. 1998;95(26):15339–44.
Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D, Shultz L, Bhatia R. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood. 2013;121(10):1824–38.
Eiring AM, Khorashad JS, Anderson DJ, Yu F, Redwine HM, Mason CC, Reynolds KR, Clair PM, Gantz KC, Zhang TY, et al. Beta-catenin is required for intrinsic but not extrinsic BCR-ABL1 kinase-independent resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia. 2015;29(12):2328–37.
Shenoy AK, Lu J. Cancer cells remodel themselves and vasculature to overcome the endothelial barrier. Cancer Lett. 2016;380(2):534–44.
Qi J, Wang J, Romanyuk O, Siu CH. Involvement of Src family kinases in N-cadherin phosphorylation and beta-catenin dissociation during transendothelial migration of melanoma cells. Mol Biol Cell. 2006;17(3):1261–72.
Zen K, Liu DQ, Guo YL, Wang C, Shan J, Fang M, Zhang CY, Liu Y. CD44v4 is a major E-selectin ligand that mediates breast cancer cell transendothelial migration. PLoS One. 2008;3(3):e1826.
Tremblay PL, Auger FA, Huot J. Regulation of transendothelial migration of colon cancer cells by E-selectin-mediated activation of p38 and ERK MAP kinases. Oncogene. 2006;25(50):6563–73.
Hu Y, Szente B, Kiely JM, Gimbrone MA Jr. Molecular events in transmembrane signaling via E-selectin. SHP2 association, adaptor protein complex formation and ERK1/2 activation. J Biol Chem. 2001;276(51):48549–53.
Zhang P, Fu C, Bai H, Song E, Dong C, Song Y. CD44 variant, but not standard CD44 isoforms, mediate disassembly of endothelial VE-cadherin junction on metastatic melanoma cells. FEBS Lett. 2014;588(24):4573–82.
Kang SA, Hasan N, Mann AP, Zheng W, Zhao L, Morris L, Zhu W, Zhao YD, Suh KS, Dooley WC, et al. Blocking the adhesion cascade at the premetastatic niche for prevention of breast cancer metastasis. Mol Ther. 2015;23(6):1044–54.
Adams GB, Scadden DT. The hematopoietic stem cell in its place. Nat Immunol. 2006;7(4):333–7.
Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6(2):93–106.
Greenbaum AM, Revollo LD, Woloszynek JR, Civitelli R, Link DC. N-cadherin in osteolineage cells is not required for maintenance of hematopoietic stem cells. Blood. 2012;120(2):295–302.
Tillet E, Vittet D, Feraud O, Moore R, Kemler R, Huber P. N-cadherin deficiency impairs pericyte recruitment, and not endothelial differentiation or sprouting, in embryonic stem cell-derived angiogenesis. Exp Cell Res. 2005;310(2):392–400.
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–8.
Petzer AL, Eaves CJ, Lansdorp PM, Ponchio L, Barnett MJ, Eaves AC. Characterization of primitive subpopulations of normal and leukemic cells present in the blood of patients with newly diagnosed as well as established chronic myeloid leukemia. Blood. 1996;88(6):2162–71.
Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322(5909):1861–5.
Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, Wagers AJ, Hsiao EC, Passegue E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285–99.
Wang X, Huang S, Chen JL. Understanding of leukemic stem cells and their clinical implications. Mol Cancer. 2017;16(1):2.
Zhou HS, Carter BZ, Andreeff M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol Med. 2016;13(2):248–59.
Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12(10):1167–74.
Hosokawa K, Arai F, Yoshihara H, Iwasaki H, Hembree M, Yin T, Nakamura Y, Gomei Y, Takubo K, Shiama H, et al. Cadherin-based adhesion is a potential target for niche manipulation to protect hematopoietic stem cells in adult bone marrow. Cell Stem Cell. 2010;6(3):194–8.
Kiel MJ, Acar M, Radice GL, Morrison SJ. Hematopoietic stem cells do not depend on N-cadherin to regulate their maintenance. Cell Stem Cell. 2009;4(2):170–9.
Bromberg O, Frisch BJ, Weber JM, Porter RL, Civitelli R, Calvi LM. Osteoblastic N-cadherin is not required for microenvironmental support and regulation of hematopoietic stem and progenitor cells. Blood. 2012;120(2):303–13.
Zhi L, Wang M, Rao Q, Yu F, Mi Y, Wang J. Enrichment of N-cadherin and Tie2-bearing CD34+/CD38-/CD123+ leukemic stem cells by chemotherapy-resistance. Cancer Lett. 2010;296(1):65–73.
Qiu S, Jia Y, Xing H, Yu T, Yu J, Yu P, Tang K, Tian Z, Wang H, Mi Y, et al. N-cadherin and Tie2 positive CD34(+)CD38(−)CD123(+) leukemic stem cell populations can develop acute myeloid leukemia more effectively in NOD/SCID mice. Leuk Res. 2014;38(5):632–7.
Zhang B, Groffen J, Heisterkamp N. Increased resistance to a farnesyltransferase inhibitor by N-cadherin expression in Bcr/Abl-P190 lymphoblastic leukemia cells. Leukemia. 2007;21(6):1189–97.
Marjon KD, Termini CM, Karlen KL, Saito-Reis C, Soria CE, Lidke KA, Gillette JM. Tetraspanin CD82 regulates bone marrow homing of acute myeloid leukemia by modulating the molecular organization of N-cadherin. Oncogene. 2016;35(31):4132–40.
Zhi L, Gao Y, Yu C, Zhang Y, Zhang B, Yang J, Yao Z. N-cadherin aided in maintaining the characteristics of leukemic stem cells. Anat Rec (Hoboken). 2016;299(7):990–8.
Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, Wang Y, Battula VL, Mak PY, Schallmoser K, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-kappaB mediates chemoresistance. Blood. 2014;123(17):2691–702.
Kurtova AV, Balakrishnan K, Chen R, Ding W, Schnabl S, Quiroga MP, Sivina M, Wierda WG, Estrov Z, Keating MJ, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. 2009;114(20):4441–50.
Hsieh YT, Gang EJ, Geng H, Park E, Huantes S, Chudziak D, Dauber K, Schaefer P, Scharman C, Shimada H, et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood. 2013;121(10):1814–8.
Latifi A, Abubaker K, Castrechini N, Ward AC, Liongue C, Dobill F, Kumar J, Thompson EW, Quinn MA, Findlay JK, et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J Cell Biochem. 2011;112(10):2850–64.
Yamauchi M, Yoshino I, Yamaguchi R, Shimamura T, Nagasaki M, Imoto S, Niida A, Koizumi F, Kohno T, Yokota J, et al. N-cadherin expression is a potential survival mechanism of gefitinib-resistant lung cancer cells. Am J Cancer Res. 2011;1(7):823–33.
Zhang X, Liu G, Kang Y, Dong Z, Qian Q, Ma X. N-cadherin expression is associated with acquisition of EMT phenotype and with enhanced invasion in erlotinib-resistant lung cancer cell lines. PLoS One. 2013;8(3):e57692.
Wu Y, Ginther C, Kim J, Mosher N, Chung S, Slamon D, Vadgama JV. Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol Cancer Res. 2012;10(12):1597–606.
Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, Chen D, Seo K, Modrusan Z, Gao WQ, et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res. 2012;72(2):527–36.
Tran NL, Adams DG, Vaillancourt RR, Heimark RL. Signal transduction from N-cadherin increases Bcl-2. Regulation of the phosphatidylinositol 3-kinase/Akt pathway by homophilic adhesion and actin cytoskeletal organization. J Biol Chem. 2002;277(36):32905–14.
Groen RW, de Rooij MF, Kocemba KA, Reijmers RM, de Haan-Kramer A, Overdijk MB, Aalders L, Rozemuller H, Martens AC, Bergsagel PL, et al. N-cadherin-mediated interaction with multiple myeloma cells inhibits osteoblast differentiation. Haematologica. 2011;96(11):1653–61.
Dring AM, Davies FE, Fenton JA, Roddam PL, Scott K, Gonzalez D, Rollinson S, Rawstron AC, Rees-Unwin KS, Li C, et al. A global expression-based analysis of the consequences of the t(4;14) translocation in myeloma. Clin Cancer Res. 2004;10(17):5692–701.
Keats JJ, Reiman T, Maxwell CA, Taylor BJ, Larratt LM, Mant MJ, Belch AR, Pilarski LM. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood. 2003;101(4):1520–9.
Keats JJ, Maxwell CA, Taylor BJ, Hendzel MJ, Chesi M, Bergsagel PL, Larratt LM, Mant MJ, Reiman T, Belch AR, et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood. 2005;105(10):4060–9.
Lauring J, Abukhdeir AM, Konishi H, Garay JP, Gustin JP, Wang Q, Arceci RJ, Matsui W, Park BH. The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood. 2008;111(2):856–64.
Ghobrial IM. Myeloma as a model for the process of metastasis: implications for therapy. Blood. 2012;120(1):20–30.
Mrozik KM, Cheong CM, Hewett D, Chow AW, Blaschuk OW, Zannettino AC, Vandyke K. Therapeutic targeting of N-cadherin is an effective treatment for multiple myeloma. Br J Haematol. 2015;171(3):387–99.
Noll JE, Williams SA, Purton LE, Zannettino AC. Tug of war in the haematopoietic stem cell niche: do myeloma plasma cells compete for the HSC niche? Blood Cancer J. 2012;2:e91.
Katz BZ. Adhesion molecules--the lifelines of multiple myeloma cells. Semin Cancer Biol. 2010;20(3):186–95.
Sadler NM, Harris BR, Metzger BA, Kirshner J. N-cadherin impedes proliferation of the multiple myeloma cancer stem cells. Am J Blood Res. 2013;3(4):271–85.
Volk T, Volberg T, Sabanay I, Geiger B. Cleavage of A-CAM by endogenous proteinases in cultured lens cells and in developing chick embryos. Dev Biol. 1990;139(2):314–26.
Harrison OJ, Corps EM, Berge T, Kilshaw PJ. The mechanism of cell adhesion by classical cadherins: the role of domain 1. J Cell Sci. 2005;118(Pt 4):711–21.
Blaschuk OW. N-cadherin antagonists as oncology therapeutics. Philos Trans R Soc B Biol Sci. 2015;370(1661):20140039.
Williams E, Williams G, Gour BJ, Blaschuk OW, Doherty P. A novel family of cyclic peptide antagonists suggests that N-cadherin specificity is determined by amino acids that flank the HAV motif. J Biol Chem. 2000;275(6):4007–12.
Shintani Y, Fukumoto Y, Chaika N, Grandgenett PM, Hollingsworth MA, Wheelock MJ, Johnson KR. ADH-1 suppresses N-cadherin-dependent pancreatic cancer progression. Int J Cancer. 2008;122(1):71–7.
Li H, Price DK, Figg WD. ADH1, an N-cadherin inhibitor, evaluated in preclinical models of angiogenesis and androgen-independent prostate cancer. Anti-Cancer Drugs. 2007;18(5):563–8.
Lammens T, Swerts K, Derycke L, De Craemer A, De Brouwer S, De Preter K, Van Roy N, Vandesompele J, Speleman F, Philippe J, et al. N-cadherin in neuroblastoma disease: expression and clinical significance. PLoS One. 2012;7(2):e31206.
Kelland L. Drug evaluation: ADH-1, an N-cadherin antagonist targeting cancer vascularization. Curr Opin Mol Ther. 2007;9(1):86–91.
Erez N, Zamir E, Gour BJ, Blaschuk OW, Geiger B. Induction of apoptosis in cultured endothelial cells by a cadherin antagonist peptide: involvement of fibroblast growth factor receptor-mediated signalling. Exp Cell Res. 2004;294(2):366–78.
Perotti A, Sessa C, Mancuso A, Noberasco C, Cresta S, Locatelli A, Carcangiu ML, Passera K, Braghetti A, Scaramuzza D, et al. Clinical and pharmacological phase I evaluation of Exherin (ADH-1), a selective anti-N-cadherin peptide in patients with N-cadherin-expressing solid tumours. Ann Oncol. 2009;20(4):741–5.
Yarom N, Stewart D, Malik R, Wells J, Avruch L, Jonker DJ. Phase I clinical trial of Exherin (ADH-1) in patients with advanced solid tumors. Curr Clin Pharmacol. 2013;8(1):81–8.
Augustine CK, Yoshimoto Y, Gupta M, Zipfel PA, Selim MA, Febbo P, Pendergast AM, Peters WP, Tyler DS. Targeting N-cadherin enhances antitumor activity of cytotoxic therapies in melanoma treatment. Cancer Res. 2008;68(10):3777–84.
Turley RS, Tokuhisa Y, Toshimitsu H, Lidsky ME, Padussis JC, Fontanella A, Deng W, Augustine CK, Beasley GM, Davies MA, et al. Targeting N-cadherin increases vascular permeability and differentially activates AKT in melanoma. Ann Surg. 2015;261(2):368–77.
Beasley GM, McMahon N, Sanders G, Augustine CK, Selim MA, Peterson B, Norris R, Peters WP, Ross MI, Tyler DS. A phase 1 study of systemic ADH-1 in combination with melphalan via isolated limb infusion in patients with locally advanced in-transit malignant melanoma. Cancer. 2009;115(20):4766–74.
Beasley GM, Riboh JC, Augustine CK, Zager JS, Hochwald SN, Grobmyer SR, Peterson B, Royal R, Ross MI, Tyler DS. Prospective multicenter phase II trial of systemic ADH-1 in combination with melphalan via isolated limb infusion in patients with advanced extremity melanoma. J Clin Oncol. 2011;29(9):1210–5.
Cosgrove BD, Mui KL, Driscoll TP, Caliari SR, Mehta KD, Assoian RK, Burdick JA, Mauck RL. N-cadherin adhesive interactions modulate matrix mechanosensing and fate commitment of mesenchymal stem cells. Nat Mater. 2016;15(12):1297–306.
Ganz A, Lambert M, Saez A, Silberzan P, Buguin A, Mege RM, Ladoux B. Traction forces exerted through N-cadherin contacts. Biol Cell. 2006;98(12):721–30.
Chopra A, Tabdanov E, Patel H, Janmey PA, Kresh JY. Cardiac myocyte remodeling mediated by N-cadherin-dependent mechanosensing. Am J Physiol Heart Circ Physiol. 2011;300(4):H1252–66.
Lee E, Ewald ML, Sedarous M, Kim T, Weyers BW, Truong RH, Yamada S. Deletion of the cytoplasmic domain of N-cadherin reduces, but does not eliminate, traction force-transmission. Biochem Biophys Res Commun. 2016;478(4):1640–6.
Su Y, Li J, Witkiewicz AK, Brennan D, Neill T, Talarico J, Radice GL. N-cadherin haploinsufficiency increases survival in a mouse model of pancreatic cancer. Oncogene. 2012;31(41):4484–9.
Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog signaling in the maintenance of Cancer stem cells. Cancers (Basel). 2015;7(3):1554–85.
Burden-Gulley SM, Gates TJ, Craig SE, Lou SF, Oblander SA, Howell S, Gupta M, Brady-Kalnay SM. Novel peptide mimetic small molecules of the HAV motif in N-cadherin inhibit N-cadherin-mediated neurite outgrowth and cell adhesion. Peptides. 2009;30(12):2380–7.
Devemy E, Blaschuk OW. Identification of a novel N-cadherin antagonist. Peptides. 2008;29(11):1853–61.
Doro F, Colombo C, Alberti C, Arosio D, Belvisi L, Casagrande C, Fanelli R, Manzoni L, Parisini E, Piarulli U, et al. Computational design of novel peptidomimetic inhibitors of cadherin homophilic interactions. Org Biomol Chem. 2015;13(9):2570–3.
Funding
This work was supported by the Cancer Australia Priority-driven Collaborative Cancer Research Scheme, co-funded by the Leukaemia Foundation. KV was supported by a research fellowship awarded by the Cancer Council SA Beat Cancer Project on behalf of its donors and the State Government of South Australia, through the Department of Health.
Author information
Authors and Affiliations
Contributions
KMM, OWB, CMC, ACW and KV contributed to manuscript conception and design. KMM performed literature search and wrote the manuscript. All authors critically edited the manuscript, and read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mrozik, K.M., Blaschuk, O.W., Cheong, C.M. et al. N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer 18, 939 (2018). https://doi.org/10.1186/s12885-018-4845-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-018-4845-0