
Abstract
Since leukemic stem cells (LSCs) or cancer stem cells (CSCs) were found in acute myeloid leukemia (AML) in 1997, extensive studies have been contributed to identification and characterization of such cell populations in various tissues. LSCs are now generally recognized as a heterogeneous cell population that possesses the capacities of self-renewal, proliferation and differentiation. It has been shown that LSCs are regulated by critical surface antigens, microenvironment, intrinsic signaling pathways, and novel molecules such as some ncRNAs. To date, significant progress has been made in understanding of LSCs, leading to the development of numerous LSCs-targeted therapies. Moreover, various novel therapeutic agents targeting LSCs are undergoing clinical trials. Here, we review current knowledge of LSCs, and discuss the potential therapies and their challenges that are being tested in clinical trials for evaluation of their effects on leukemias.
Similar content being viewed by others


Background
The existence of CSCs was firstly evidenced in AML [1], and has been now extended to a broad spectrum of solid tumors [2–8]. In 1994, Dick and colleagues [1] showed that only the leukemic cells expressing the same markers as normal adult hematopoietic stem cells (CD34+CD38−) could initiate hematopoietic malignancy, and termed these cells as leukemia-initiating cells, leukemic stem cells (LSCs), or cancer stem cells (CSCs) [1, 9, 10]. Importantly, such cell population possesses the capacities for self-renewal, proliferation and differentiation. Increasing evidence has demonstrated that LSCs are clinically relevant, indicating that therapies targeting LSCs in AML would improve survival outcomes [11].
Conventional anticancer strategy is a combination of surgery, chemotherapy and radiotherapy with allogeneic stem cell transplantation for eligible candidates [12–14]. However, elderly patients cannot tolerate such intense regimens, and patients usually face the risk of recurrence, metastasis and drug resistance. It is thought that these therapies predominantly target at a bulk tumor populations but leave CSCs behind. Importantly, these CSCs, with highly expressed ATP-binding cassette (ABC) transporters, have been shown to protect themselves from the attacks from chemotherapeutic agents [15–17]. Hence, the inefficient therapy of cancers is mainly attributed to the failure of elimination of the malignant CSCs. It is well recognized that development of CSC-selective therapies is important for treating CSCs-containing cancers [18]. In this review, we discuss the current understanding of LSCs. Also, we summarize various therapeutic agents targeting LSCs that are being studied in clinical trials.
Genetic and epigenetic heterogeneities of LSCs
Leukemias are now viewed as aberrant hematopoietic processes initiated by rare LSCs, which arise from the transformation of hematopoietic stem cells (HSCs) or committed progenitor cells [19]. During the course of malignant transformation, LSCs acquire the capacity of self-renewal, proliferation and differentiation through continuous genetic and epigenetic alteration and clonal diversification. Thus, understanding how genetic and epigenetic heterogeneities develop in different leukemias has become an important area for cancer research. Although CSCs have been found in both leukemia and solid tumors, not all of CSCs in the solid tumors follow the heterogeneity model of LSC.
Increasing investigations using deep genome sequencing have identified many recurrent mutated genes critically implicated in the pathogenesis of human AML [20–27]. In 2013, the Cancer Genome Atlas Research Network analyzed the genome of 200 AML patients, and thoroughly defined the recurrent mutations in AML [28]. About 30 genes were identified to be mutated in more than 2% of patients. Remarkably, many of these mutated genes encode proteins that normally function at the epigenetic level, including modifications of DNA cytosine residues and post-translational modifications of histones. In addition, other studies have shown that clonal composition of AMLs appears to be changed quite markedly at both the genetic and epigenetic levels after therapy in relapsed disease [29–31].
Interestingly, it has been found that there is a sequential order for the acquisition of these mutations during leukemogenesis. For example, some researchers observed that somatic mutations in epigenetic modifiers that regulate cytosine methylation, such as DNMT3A (DNA methyltransferase 3 alpha), IDH1/2 (isocitrate dehydrogenase 1/2) and TET2 (tet methylcytosine dioxygenase 2), occur early in pre-leukemic HSCs [32–34]. However, other somatic mutations in signaling pathways that drive proliferation, such as NPM1 (nucleophosmin 1), FLT3-ITD (internal tandem duplication of the gene FLT3) and KRAS/NRAS (Kirsten rat sarcoma viral oncogene homolog/neuroblastoma rat sarcoma viral oncogene homolog), are later events in AML transformation [35]. These results suggest that disruption of epigenetic patterning is likely an early and prominent event during leukemogenesis.
In order to characterize the expression profile of LSCs in chronic myeloid leukemia (CML), Gerber and colleagues performed genome-wide transcriptome analysis of CML LSCs using exon microarrays [36]. They identified 97 genes that are differentially expressed between CML LSCs and normal HSCs. Further analysis revealed dysregulation of proliferation, differentiation and signaling pathways in CML LSCs. These data may provide potential therapeutic targets unique to CML LSCs.
Surface molecules and microenvironment of LSCs and their clinical implications
Cell surface molecules of LSCs
The AML LSCs were the first reported and best characterized type of CSCs, and they specifically display CD34+CD38− cell surface markers [1, 9, 10]. However, subsequent studies showed that the surface markers of AML LSCs are considerably heterogeneous [37–47]. For example, AML LSCs were found not only in Lin−/CD38− fractions but also in CD34−, Lin+, CD38+, and CD45RA+ fractions [45]. It was also found that true AML LSCs in the CD34+/CD38− fractions, originally described by Bonnet and Dick, were very rare and comprised a hierarchy of cells with different self-renewal potential [46]. In addition, some surface markers of AML LSCs (CD34+, CD38−, CD71−, and HLA-DR−) are shared with normal HSCs, and others (Lin+, CD38+, CD45RA+) are associated with normal committed progenitors [38, 45]. These findings stirred up a debate about whether AML LSCs are derived from the normal HSCs or from the committed progenitor cells. On the other hand, the surface markers of LSCs are heterogeneous, which makes hard for classification of LSCs and even LSCs-targeted treatment in clinics.
Recently, great progress has been made in understanding of LSC surface markers and their clinical applications, especially in AML cases. Firstly, a number of critical surface markers unique to AML LSCs have been identified. For example, it has been revealed that CD90 and CD117 are deficient in AML LSCs [39], while CD123 [42, 48], TIM3 [44, 49], CD47 [50, 51], CD96 [52], CLL-1 [53, 54], and IL-1 receptor accessory protein (IL1RAP) [55] are highly expressed in AML LSCs. Targeting these surface markers is a promising strategy for eradicating AML LSCs. Previous studies have shown that CD123 (IL-3 receptor α chain) was preferentially expressed in the CD34+/CD38− AML cells, as compared with normal HSC samples. Pretreatment of AML cells with anti-CD123 monoclonal antibody 7G3 resulted in decreased engraftment when they were injected into a xenograft model [42, 48]. To date, phase I clinical trials (NCT00401739 and NCT01632852) of using monoclonal antibody targeting CD123 (CSL360 and improved CSL362) [48] have been tested in CD123+ AML patients. Moreover, other monoclonal antibodies targeting CD47 [56, 57], CD96 [52, 58], TIM3 [44, 49] and CLL-1 [54, 59] have also been investigated in pre-clinical models for their ability to eliminate primary AML LSCs. It is worth mentioning that Gemtuzumab Ozogamicin, an anti-CD33 antibody, is the first monoclonal antibody approved by the Food and Drug Administration (FDA) of the USA in 2000 for the treatment of AML, although it may not specifically target LSCs [60].
Secondly, increasing novel therapies are continuously developed to specifically target these surface antigens of LSCs and are undergoing in clinical trials in AML cases. Besides monoclonal antibodies mentioned above [61, 62], these new therapies include both bi-specific and tri-specific antibody fragments [63, 64], immunotoxins [65], chimeric antigen receptor modified T-cells (CAR T-cells) [66], and nano-particles containing surface markers-targeted medication [67]. Notably, DT388IL3 (SL-401) is a recombinant immunotoxin that is created by fusing diphtheria toxin with a ligand targeting IL-3 receptor. At present, DT388IL3 (SL-401) undergoes phase I/II trials (NCT02113982 and NCT02270463) in AML [65] (Table 1).
Microenvironment associated with LSCs
Under normal conditions, HSCs rely on the interactions with the bone marrow (BM) niche, which is critical for their proper function and maintenance [68]. The remodeling of the BM niche is commonly observed in blood malignancies. There is evidence that growth of leukemic cells disrupts the BM niches of normal hematopoietic progenitor cells and creates a microenvironment hospitable for them [69]. Within such microenvironment, LSCs are able to communicate with BM stromal cells through cytokines, chemokines and intracellular signals initiated by cellular adhesion [70, 71]. Importantly, these signals influence the ability of LSCs to self-renew, maintain their quiescence, and prevent apoptosis. In addition, the BM niche provides two distinct microenvironmental zones (the osteoblastic niche and vascular niche) that likely regulate the cycling of LSCs [71–73]. Thus, blocking the interactions between LSCs and their microenvironment represents a promising strategy to disrupt LSC homeostasis and restore normal hematopoiesis.
One of such strategies is to dislodge LSCs from their protective BM niche and thus sensitize the LSCs to conventional chemotherapies. It has been demonstrated that LSCs migrate into and remain within the BM niche through the interaction between C-X-C chemokine receptor type 4 (CXCR4) and stromal cell derived factor-1 (SDF-1α), also known as C-X-C motif chemokine 12 (CXCL12) [74]. Recently, manipulating the CXCL12-CXCR4 axis using Plerixafor (AMD3100) in relapsed AML has been reported as a safe strategy in phase I/II clinical trials (NCT00990054 and NCT00822770) [75–78]. Additionally, ligation of the adhesion molecules CD44 [79] and vascular cell adhesion molecule 1 (VCAM-1) [80] with their monoclonal antibodies has already been tested in the clinic. Other strategies like altering BM niche remodeling and inflammatory microenvironment, such as targeting pro-inflammatory cytokines tumor necrosis factor alpha (TNFα), IL-1, and IL-6, might be very promising but mainly at pre-clinical stages [81].
Intracellular molecules and signaling of LSCs
Critical signaling pathways involved in regulation of LSCs
LSCs are characterized by limitless self-renewal, proliferation and differentiation. A set of critical genes impact these functional properties through a wide range of cellular pathways and processes, which have been described in detail by many groups [13, 19, 71, 82]. Signaling pathways such as Wnt/β-catenin [83–89] and Hedgehog [90–92] play important role in regulating self-renewal of LSCs. These signaling pathways are also critically required for the development of normal HSCs [93]. In addition, it is thought that LSCs can evade apoptosis by up-regulating NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) [94, 95] or by down-regulating Fas/CD95 [96]. Here, we review some key signaling pathways involved in the regulation of survival and self-renewal of LSCs.
The well-known Wnt/β-catenin signaling pathway plays fundamental role in maintaining CSC populations. The activation of Wnt/β-catenin pathway leads to the translocation of β-catenin into the nucleus, where it induces the expression of target genes such as c-Myc, c-Jun and cyclin D1 [97–101]. Various experiments have demonstrated that Wnt/β-catenin signaling pathway acts as a key regulator in controlling proliferation, survival, and differentiation of hematopoietic cells [99, 102]. Aberrant activation of Wnt/β-catenin signaling pathway has also been found in both AML [87–89] and CML [83, 84]. Subsequent studies have shown that Wnt/β-catenin signaling pathway is required for efficient self-renewal of LSCs, indicating that it is an attractive therapeutic strategy to target Wnt/β-catenin signaling in AML and CML [84, 85]. In addition, it has been documented that Wnt signaling pathway and the polycomb-group protein BMI1 (B lymphoma Mo-MLV insertion region 1 homolog) are involved in the expansion of LSCs [103–105].
Janus kinase (JAK)/signal transducer and activator of transcription (STAT) and phosphatidylinositide 3-kinase (PI3K)/protein kinase B (AKT) are two crucial signaling pathways that have been implicated in LSC survival and multiple cancer formation. It is well established that malignant transformation of many cell types, especially hematopoietic cells, involves the dysregulation of JAK/STAT and/or PI3K/AKT that regulate cellular proliferation and survival. For example, there is considerable evidence showing that aberrations in these signaling pathways are associated with numerous leukemias. In CML, JAK/STAT/PIM (proviral insertion in murine) and PI3K/AKT/mTOR (mammalian/mechanistic target of rapamycin) pathways are constitutively activated by Bcr-Abl, a non-receptor tyrosine kinase, resulting in uncontrolled cellular proliferation [12, 106–108]. Bcr-Abl can also cause tyrosine phosphorylation of suppressors of cytokine signaling 1 and 3 (SOCS-1 and SOCS-3), two potent suppressors of JAK/STAT signaling, and thereby diminish their inhibitory effects on JAK/STAT activation [109]. Interestingly, PI3K mutation and AKT1 (E17K) mutation has been identified in a variety of tumors. AKT1 (E17K) mutant, a constitutively activated form of AKT1, can significantly promote tumorigenesis [110]. In addition, it was observed that other members of the PI3K/AKT/mTOR pathway, such as PTEN (phosphatase and tensin homolog) and mTOR, function in the maintenance of LSCs [111]. Recently, we have shown that there exists a crosstalk between JAK/STAT/PIM and PI3K/AKT/mTOR pathways that converge on eukaryotic translation initiation factor 4B (eIF4B) to regulate the survival of Abl transformants [112, 113].
In brief, increasing evidence has suggested that multiple signaling pathways are involved in the development of LSCs. Profound elucidation of the intricate pathway network in LSCs is significant in understanding of LSCs and designing precise treatment of leukemia through targeting LSCs. Currently, various clinical trials are in process to test the efficacy of agents targeting intracellular proteins and pathways associated with LSCs. For example, clinical studies of the drug CAL-101, an inhibitor of PI3K, showed remarkable success in chronic lymphocytic leukemia (CLL). It has also been found that CAL-101 has some effects on tumor microenvironment [114]. Additionally, other inhibitors targeting PI3K/AKT/mTOR, NF-κB and Wnt signaling in the clinic are listed in Table 1.
Functional involvement of non-coding RNAs in malignant hematopoiesis
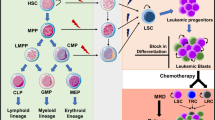
Non-coding RNAs (ncRNAs), such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), play critical roles in multiple biological processes [115–119]. Aberrant expression and functioning of these ncRNAs have been shown to be associated with various cancers and cancer stem cells [120–125]. Here, we highlight several miRNAs and lncRNAs as key regulators in hematopoietic cells and LSCs (Fig. 1).

Involvement of miRNAs and lncRNAs in normal and malignant hematopoiesis. miRNAs and lncRNAs regulate almost every step of development and differentiation of hematopoietic cells during both normal and malignant hematopoiesis. Dysregulation of the ncRNAs (in red color) is associated with transformation of hematopoietic cells
Regulation of hematopoietic malignancies by miRNAs
miRNAs are 18–22 nucleotides ncRNAs that generally regulate gene expression by promoting mRNA degradation or inhibiting mRNA translation [126, 127]. During the tumorigenesis, some miRNAs act as oncogenes, whereas others function as tumor suppressors [128–132]. They can regulate cell growth, proliferation, survival, migration and invasion of cancer cells. Notably, the roles of well-known miRNAs in normal and malignant hematopoiesis have been extensively reviewed [133–137]. These miRNAs regulate almost every step of development and differentiation of both normal hematopoietic cells and LSCs.
MiR-125 is a highly conserved miRNA. There are three homologs of miR-125 (hsa-miR-125b-1, hsa-miR-125b-2 and hsa-miR-125a) in human [136]. Previous investigations have revealed that highly expressed miR-125 enhances self-renewal and survival of HSCs, and dysregulation of miR-125 occurs in multiple hematopoietic malignancies [138–142]. In particular, miR-125 is implicated in hematopoiesis through the p53-involved regulation network [143].
Recently, Lechman and colleagues have shown that miR-126 preserves AML LSC quiescence and promotes chemotherapy resistance by targeting the PI3K/AKT/mTOR signaling pathway [144]. Interestingly, reduction of miR-126 level impairs LSC maintenance, but it plays an opposing role in normal HSCs [144]. In addition, functional involvement of miR-29a has also been found in AML LSCs [134]. Previous experiments demonstrated that miR-29a was highly expressed in AML samples. Furthermore, results exhibited that miR-29a can promote proliferation of hematopoietic progenitor, and transform AML by converting myeloid progenitors into LSCs [134].
Involvement of lncRNAs in leukemogenesis
Over the past decade, increasing numbers of lncRNAs have been identified and recognized as novel regulators that are implicated in various cellular processes. LncRNAs are generally more than 200 nucleotides in length, and modulate gene expression through interaction with DNAs, RNAs and proteins [145–147]. They function at multiple levels, including gene transcription, post-transcriptional processing, RNA translation, and epigenetic modifications [148]. It has been reported that some lncRNAs are involved in the regulation of CSCs [149–152]. For example, the lncRNA, named lncTCF7, has been identified to promote liver CSC self-renewal and tumor propagation by activating Wnt signaling [149]. Moreover, many lncRNAs have been seen to be associated with normal hematopoietic cells and various types of leukemia [118, 153–160] (Fig. 1).
Dysregulation of lncRNA H19 has been observed in various tumors, including Bcr-Abl-induced leukemia [161–164]. H19 acts as dual regulators in different cancers (either as an oncogene or a tumor suppressor) and also serves as a precursor for miR-675, known to down-regulate the tumor suppressor gene RB in human colorectal cancer [165–167]. Importantly, H19 is highly expressed in long-term HSCs (LT-HSCs). H19-deficiency results in activation of the insulin-like growth factor 2 (IGF2)-IGF1 receptor pathway, leading to increased proliferation and decreased long-term self-renewal of HSCs [168].
Recently, Guo et al. have comprehensively analyzed the expression of lncRNAs in human CML cells [158]. Notably, a lncRNA termed lncRNA-BGL3 was highly induced in response to silence of Bcr-Abl expression or inhibition of Bcr-Abl kinase activity in K562 cells and leukemic cells derived from CML patients. Furthermore, lncRNA-BGL3 functions as a competitive endogenous RNA (ceRNA) to cross-regulate PTEN expression, thereby modulating leukemic cell survival. Thus, lncRNA-BGL3 has been identified as a tumor suppressor in Bcr-Abl-mediated cellular transformation.
To date, miRNAs and lncRNAs have been confirmed by increasing evidence as functional mediators in cancer cells and cancer stem cells. Some cancer-associated ncRNAs are currently considered as biomarkers for patient prognosis and potential therapeutic agents for particular cancers [128, 129, 169–181]. For example, MRX34, the first miRNA mimic, entered phase I clinical trials in patients with advanced hepatocellular carcinoma in 2013 [169]. In AML, Dorrance et al. have observed that miR-126 enriches in AML LSCs and contributes to the long-term maintenance and self-renewal of LSCs. Treatment with novel nano-particles containing antagomiR-126 results in reduction of LSCs in vivo [181]. Therefore, better understanding of the mechanisms underlying functional involvement of miRNAs and lncRNAs in LSC development and leukemogenesis is of great importance for precise treatment of hematopoietic malignancies.
Conclusion
Over the past two decades, the function and phenotype of LSCs have been continuously defined. Furthermore, numerous studies provide accumulating evidence that there exist CSCs in a variety of solid tumors [182, 183]. Importantly, these progresses have led to the development of many novel therapeutic strategies targeting CSCs. Here, we have reviewed the current understanding of LSCs both in intrinsic and extrinsic aspects, and discussed the promising therapeutics that is being tested in clinical trials. Although identification and characterization of LSCs have renewed leukemia research and helped develop diverse clinical therapeutic strategies, some tough challenges for LSCs-based leukemia therapy still remain. One of the greatest challenges is early and efficient identification of LSCs in diverse leukemia patients. Moreover, better understanding of LSCs development and differentiation is critically required for clinical implications of the strategies targeting such cell populations. Precise mechanisms by which extracellular and intracellular molecules and their signaling regulate LSCs also remain to be determined. Therefore, further efforts are needed to identify more specific biomarkers of LSCs, determine specific targets and thereby develop efficient LSCs-based treatment of leukemia.
Abbreviations
- ABC transporters:
-
ATP-binding cassette transporters
- AML:
-
Acute myeloid leukemia
- BM niche:
-
Bone marrow niche
- BMI1:
-
B lymphoma Mo-MLV insertion region 1 homolog
- CAR T-cells:
-
Chimeric antigen receptor modified T-cells
- CEBPE:
-
CCAAT/enhancer binding protein epsilon
- CEPBA:
-
CCAAT/enhancer binding protein alpha
- CeRNA:
-
Competitive endogenous RNA
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- CSCs:
-
Cancer stem cells
- CXCL12:
-
C-X-C motif chemokine 12
- CXCR4:
-
C-X-C chemokine receptor type 4
- DNMT3A :
-
DNA methyltransferase 3 alpha
- eIF4B:
-
Eukaryotic translation initiation factor 4B
- FDA:
-
Food and Drug Administration
- FLT3-ITD :
-
Internal tandem duplication of the gene FLT3
- HSCs:
-
Hematopoietic stem cells
- IDH1/2 :
-
Isocitrate dehydrogenase 1/2
- IGF2:
-
Insulin-like growth factor 2
- IL1RAP:
-
IL-1 receptor accessory protein
- JAK/STAT:
-
Janus kinase/signal transducer and activator of transcription
- KRAS/NRAS:
-
Kirsten rat sarcoma viral oncogene homolog/neuroblastoma rat sarcoma viral oncogene homolog
- lncRNAs:
-
Long non-coding RNAs
- LSCs:
-
Leukemic stem cells
- LT-HSCs:
-
Long-term HSCs
- miRNAs:
-
microRNAs
- mTOR:
-
mammalian/mechanistic target of rapamycin
- ncRNAs:
-
non-coding RNAs
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NPM1:
-
Nucleophosmin 1
- PI3K/AKT:
-
Phosphatidylinositide 3-kinase/protein kinase B
- PIM:
-
Proviral insertion in murine
- PTEN:
-
Phosphatase and tensin homolog
- SDF-1α:
-
Stromal cell derived factor-1
- SOCS-1 and SOCS-3:
-
suppressors of cytokine signaling 1 and 3
- TET2:
-
Tet methylcytosine dioxygenase 2
- TNFα:
-
Tumor necrosis factor alpha
- VCAM-1:
-
Vascular cell adhesion molecule 1
References
Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8.
Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8.
Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–35.
Burns JS, Abdallah BM, Guldberg P, et al. Tumorigenic heterogeneity in cancer stem cells evolved from long-term cultures of telomerase-immortalized human mesenchymal stem cells. Cancer Res. 2005;65:3126–35.
Collins AT, Berry PA, Hyde C, et al. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–51.
Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci U S A. 2006;103:11154–9.
Piccirillo SG, Reynolds BA, Zanetti N, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–5.
O’Brien CA, Pollett A, Gallinger S, et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10.
Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7.
Bhatia M, Wang JC, Kapp U, et al. Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci U S A. 1997;94:5320–5.
Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–93.
Apperley JF. Chronic myeloid leukaemia. Lancet. 2015;385:1447–59.
Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34:732–40.
Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373:1136–52.
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–84.
Matsui W, Wang Q, Barber JP, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008;68:190–7.
Raaijmakers MH. ATP-binding-cassette transporters in hematopoietic stem cells and their utility as therapeutical targets in acute and chronic myeloid leukemia. Leukemia. 2007;21:2094–102.
Carnero A, Garcia-Mayea Y, Mir C, et al. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat Rev. 2016;49:25–36.
Lane SW, Gilliland DG. Leukemia stem cells. Semin Cancer Biol. 2010;20:71–6.
Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72.
Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–7.
Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66.
Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33.
Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–6.
Nikoloski G, Langemeijer SM, Kuiper RP, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42:665–7.
Greif PA, Eck SH, Konstandin NP, et al. Identification of recurring tumor-specific somatic mutations in acute myeloid leukemia by transcriptome sequencing. Leukemia. 2011;25:821–7.
Yan XJ, Xu J, Gu ZH, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–15.
Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10.
Kronke J, Bullinger L, Teleanu V, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122:100–8.
Li S, Garrett-Bakelman F, Perl AE, et al. Dynamic evolution of clonal epialleles revealed by methclone. Genome Biol. 2014;15:472.
Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–78.
Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–33.
Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–8.
Corces-Zimmerman MR, Hong WJ, Weissman IL, et al. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A. 2014;111:2548–53.
Gerber JM, Gucwa JL, Esopi D, et al. Genome-wide comparison of the transcriptomes of highly enriched normal and chronic myeloid leukemia stem and progenitor cell populations. Oncotarget. 2013;4:715–28.
Blair A, Hogge DE, Ailles LE, et al. Lack of expression of Thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood. 1997;89:3104–12.
Blair A, Hogge DE, Sutherland HJ. Most acute myeloid leukemia progenitor cells with long-term proliferative ability in vitro and in vivo have the phenotype CD34(+)/CD71(−)/HLA-DR. Blood. 1998;92:4325–35.
Blair A, Sutherland HJ. Primitive acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo lack surface expression of c-kit (CD117). Exp Hematol. 2000;28:660–71.
Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14:1777–84.
Taussig DC, Miraki-Moud F, Anjos-Afonso F, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–75.
Jin L, Lee EM, Ramshaw HS, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5:31–42.
Taussig DC, Vargaftig J, Miraki-Moud F, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(−) fraction. Blood. 2010;115:1976–84.
Kikushige Y, Shima T, Takayanagi S, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7:708–17.
Sarry JE, Murphy K, Perry R, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac-deficient mice. J Clin Invest. 2011;121:384–95.
Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–43.
Moshaver B, van Rhenen A, Kelder A, et al. Identification of a small subpopulation of candidate leukemia-initiating cells in the side population of patients with acute myeloid leukemia. Stem Cells. 2008;26:3059–67.
Busfield SJ, Biondo M, Wong M, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia. 2014;28:2213–21.
Jan M, Chao MP, Cha AC, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci U S A. 2011;108:5009–14.
Jaiswal S, Jamieson CH, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–85.
Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–99.
Hosen N, Park CY, Tatsumi N, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A. 2007;104:11008–13.
van Rhenen A, van Dongen GA, Kelder A, et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110:2659–66.
Zhao X, Singh S, Pardoux C, et al. Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica. 2010;95:71–8.
Askmyr M, Agerstam H, Hansen N, et al. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood. 2013;121:3709–13.
Weiskopf K, Ring AM, Ho CC, et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341:88–91.
Theocharides AP, Jin L, Cheng PY, et al. Disruption of SIRPalpha signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J Exp Med. 2012;209:1883–99.
Mohseni Nodehi S, Repp R, Kellner C, et al. Enhanced ADCC activity of affinity maturated and Fc-engineered mini-antibodies directed against the AML stem cell antigen CD96. PLoS One. 2012;7:e42426.
Bakker AB, van den Oudenrijn S, Bakker AQ, et al. C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004;64:8443–50.
Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7:1490–6.
Majeti R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene. 2011;30:1009–19.
Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–27.
Kuo SR, Wong L, Liu JS. Engineering a CD123xCD3 bispecific scFv immunofusion for the treatment of leukemia and elimination of leukemia stem cells. Protein Eng Des Sel. 2012;25:561–9.
Kugler M, Stein C, Kellner C, et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br J Haematol. 2010;150:574–86.
Frankel A, Liu JS, Rizzieri D, et al. Phase I clinical study of diphtheria toxin-interleukin 3 fusion protein in patients with acute myeloid leukemia and myelodysplasia. Leuk Lymphoma. 2008;49:543–53.
Tettamanti S, Marin V, Pizzitola I, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161:389–401.
Kievit FM, Zhang M. Cancer nanotheranostics: improving imaging and therapy by targeted delivery across biological barriers. Adv Mater. 2011;23:H217–47.
Schepers K, Campbell TB, Passegue E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell. 2015;16:254–67.
Colmone A, Amorim M, Pontier AL, et al. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322:1861–5.
Roboz GJ, Guzman M. Acute myeloid leukemia stem cells: seek and destroy. Expert Rev Hematol. 2009;2:663–72.
Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol. 2011;29:591–9.
Perry JM, Li L. Disrupting the stem cell niche: good seeds in bad soil. Cell. 2007;129:1045–7.
Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–41.
Spoo AC, Lubbert M, Wierda WG, et al. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood. 2007;109:786–91.
Uy GL, Rettig MP, Motabi IH, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917–24.
Broxmeyer HE, Orschell CM, Clapp DW, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–18.
Mohty M, Duarte RF, Croockewit S, et al. The role of plerixafor in optimizing peripheral blood stem cell mobilization for autologous stem cell transplantation. Leukemia. 2011;25:1–6.
Nervi B, Ramirez P, Rettig MP, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113:6206–14.
Krause DS, Lazarides K, von Andrian UH, et al. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. 2006;12:1175–80.
Jacamo R, Chen Y, Wang Z, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-kappaB mediates chemoresistance. Blood. 2014;123:2691–702.
Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13:285–99.
Chan WI, Huntly BJ. Leukemia stem cells in acute myeloid leukemia. Semin Oncol. 2008;35:326–35.
Zhao C, Blum J, Chen A, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12:528–41.
Hu Y, Chen Y, Douglas L, et al. beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia. 2009;23:109–16.
Wang Y, Krivtsov AV, Sinha AU, et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–3.
Yeung J, Esposito MT, Gandillet A, et al. beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell. 2010;18:606–18.
Griffiths EA, Gore SD, Hooker C, et al. Acute myeloid leukemia is characterized by Wnt pathway inhibitor promoter hypermethylation. Leuk Lymphoma. 2010;51:1711–9.
Gandillet A, Park S, Lassailly F, et al. Heterogeneous sensitivity of human acute myeloid leukemia to beta-catenin down-modulation. Leukemia. 2011;25:770–80.
Siapati EK, Papadaki M, Kozaou Z, et al. Proliferation and bone marrow engraftment of AML blasts is dependent on beta-catenin signalling. Br J Haematol. 2011;152:164–74.
Dierks C, Beigi R, Guo GR, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14:238–49.
Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–9.
Jagani Z, Dorsch M, Warmuth M. Hedgehog pathway activation in chronic myeloid leukemia. Cell Cycle. 2010;9:3449–56.
Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11.
Guzman ML, Neering SJ, Upchurch D, et al. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98:2301–7.
Guzman ML, Rossi RM, Karnischky L, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163–9.
Costello RT, Mallet F, Gaugler B, et al. Human acute myeloid leukemia CD34+/CD38-progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000;60:4403–11.
Reya T, Duncan AW, Ailles L, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–14.
Li VS, Ng SS, Boersema PJ, et al. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell. 2012;149:1245–56.
Cobas M, Wilson A, Ernst B, et al. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med. 2004;199:221–9.
Jeannet G, Scheller M, Scarpellino L, et al. Long-term, multilineage hematopoiesis occurs in the combined absence of beta-catenin and gamma-catenin. Blood. 2008;111:142–9.
Staal FJ, Clevers HC. WNT signalling and haematopoiesis: a WNT-WNT situation. Nat Rev Immunol. 2005;5:21–30.
Duchartre Y, Kim YM, Kahn M. The Wnt signaling pathway in cancer. Crit Rev Oncol Hematol. 2016;99:141–9.
Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–60.
Park IK, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–5.
Schuringa JJ, Vellenga E. Role of the polycomb group gene BMI1 in normal and leukemic hematopoietic stem and progenitor cells. Curr Opin Hematol. 2010;17:294–9.
Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164–72.
Quintas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113:1619–30.
Chen JL, Limnander A, Rothman PB. Pim-1 and Pim-2 kinases are required for efficient pre-B-cell transformation by v-Abl oncogene. Blood. 2008;111:1677–85.
Qiu X, Guo G, Chen K, et al. A requirement for SOCS-1 and SOCS-3 phosphorylation in Bcr-Abl-induced tumorigenesis. Neoplasia. 2012;14:547–58.
Guo G, Qiu X, Wang S, et al. Oncogenic E17K mutation in the pleckstrin homology domain of AKT1 promotes v-Abl-mediated pre-B-cell transformation and survival of Pim-deficient cells. Oncogene. 2010;29:3845–53.
Gutierrez A, Sanda T, Grebliunaite R, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114:647–50.
Yang J, Wang J, Chen K, et al. eIF4B phosphorylation by pim kinases plays a critical role in cellular transformation by Abl oncogenes. Cancer Res. 2013;73:4898–908.
Chen K, Yang J, Li J, et al. eIF4B is a convergent target and critical effector of oncogenic Pim and PI3K/Akt/mTOR signaling pathways in Abl transformants. Oncotarget. 2016;7:10073–89.
Fruman DA, Rommel C. PI3Kdelta inhibitors in cancer: rationale and serendipity merge in the clinic. Cancer Discov. 2011;1:562–72.
Paralkar VR, Weiss MJ. Long noncoding RNAs in biology and hematopoiesis. Blood. 2013;121:4842–6.
Ng SY, Lin L, Soh BS, et al. Long noncoding RNAs in development and disease of the central nervous system. Trends Genet. 2013;29:461–8.
Atianand MK, Fitzgerald KA. Long non-coding RNAs and control of gene expression in the immune system. Trends Mol Med. 2014;20:623–31.
Satpathy AT, Chang HY. Long noncoding RNA in hematopoiesis and immunity. Immunity. 2015;42:792–804.
Ouyang J, Zhu X, Chen Y, et al. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe. 2014;16:616–26.
Undi RB, Kandi R, Gutti RK. MicroRNAs as Haematopoiesis Regulators. Adv Hematol. 2013;2013:695754.
Croce CM. MicroRNA dysregulation in acute myeloid leukemia. J Clin Oncol. 2013;31:2065–6.
Machova Polakova K, Lopotova T, Klamova H, et al. Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol Cancer. 2011;10:41.
Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–61.
Martens-Uzunova ES, Bottcher R, Croce CM, et al. Long noncoding RNA in prostate, bladder, and kidney cancer. Eur Urol. 2014;65:1140–51.
Takahashi K, Yan I, Haga H, et al. Long noncoding RNA in liver diseases. Hepatology. 2014;60:744–53.
Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321–33.
Gregory RI, Shiekhattar R. MicroRNA biogenesis and cancer. Cancer Res. 2005;65:3509–12.
Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460–9.
Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov. 2013;12:847–65.
Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66.
Nicoloso MS, Spizzo R, Shimizu M, et al. MicroRNAs-the micro steering wheel of tumour metastases. Nat Rev Cancer. 2009;9:293–302.
Yendamuri S, Calin GA. The role of microRNA in human leukemia: a review. Leukemia. 2009;23:1257–63.
Han YC, Park CY, Bhagat G, et al. microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J Exp Med. 2010;207:475–89.
Zimmerman AL, Wu S. MicroRNAs, cancer and cancer stem cells. Cancer Lett. 2011;300:10–9.
Shaham L, Binder V, Gefen N, et al. MiR-125 in normal and malignant hematopoiesis. Leukemia. 2012;26:2011–8.
Morris VA, Zhang A, Yang T, et al. MicroRNA-150 expression induces myeloid differentiation of human acute leukemia cells and normal hematopoietic progenitors. PLoS One. 2013;8:e75815.
Enomoto Y, Kitaura J, Hatakeyama K, et al. Emu/miR-125b transgenic mice develop lethal B-cell malignancies. Leukemia. 2011;25:1849–56.
Chapiro E, Russell LJ, Struski S, et al. A new recurrent translocation t(11;14)(q24;q32) involving IGH@ and miR-125b-1 in B-cell progenitor acute lymphoblastic leukemia. Leukemia. 2010;24:1362–4.
Klusmann JH, Li Z, Bohmer K, et al. miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukemia. Genes Dev. 2010;24:478–90.
Bousquet M, Quelen C, Rosati R, et al. Myeloid cell differentiation arrest by miR-125b-1 in myelodysplastic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. J Exp Med. 2008;205:2499–506.
Zhou Y, Chen L, Barlogie B, et al. High-risk myeloma is associated with global elevation of miRNAs and overexpression of EIF2C2/AGO2. Proc Natl Acad Sci U S A. 2010;107:7904–9.
Tarasov V, Jung P, Verdoodt B, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–93.
Lechman ER, Gentner B, Ng SW, et al. miR-126 Regulates Distinct Self-Renewal Outcomes in Normal and Malignant Hematopoietic Stem Cells. Cancer Cell. 2016;29:214–28.
Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–9.
Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494–504.
Bonasio R, Shiekhattar R. Regulation of transcription by long noncoding RNAs. Annu Rev Genet. 2014;48:433–55.
Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. 2013;20:300–7.
Wang Y, He L, Du Y, et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell. 2015;16:413–25.
Zhu P, Wang Y, Huang G, et al. lnc-beta-Catm elicits EZH2-dependent beta-catenin stabilization and sustains liver CSC self-renewal. Nat Struct Mol Biol. 2016;23:631–9.
Wang X, Sun W, Shen W, et al. Long non-coding RNA DILC regulates liver cancer stem cells via IL-6/STAT3 axis. J Hepatol. 2016;64:1283–94.
Zhou X, Gao Q, Wang J, et al. Linc-RNA-RoR acts as a “sponge” against mediation of the differentiation of endometrial cancer stem cells by microRNA-145. Gynecol Oncol. 2014;133:333–9.
Sehgal L, Mathur R, Braun FK, et al. FAS-antisense 1 lncRNA and production of soluble versus membrane Fas in B-cell lymphoma. Leukemia. 2014;28:2376–87.
Trimarchi T, Bilal E, Ntziachristos P, et al. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell. 2014;158:593–606.
Sun J, Li W, Sun Y, et al. A novel antisense long noncoding RNA within the IGF1R gene locus is imprinted in hematopoietic malignancies. Nucleic Acids Res. 2014;42:9588–601.
Hughes JM, Legnini I, Salvatori B, et al. C/EBPalpha-p30 protein induces expression of the oncogenic long non-coding RNA UCA1 in acute myeloid leukemia. Oncotarget. 2015;6:18534–44.
Wang X, Chen K, Guo G, et al. Noncoding RNAs and their functional involvement in regulation of chronic myeloid leukemia. Brief Funct Genomics. 2016;15:239–48.
Guo G, Kang Q, Zhu X, et al. A long noncoding RNA critically regulates Bcr-Abl-mediated cellular transformation by acting as a competitive endogenous RNA. Oncogene. 2015;34:1768–79.
Nobili L, Lionetti M, Neri A. Long non-coding RNAs in normal and malignant hematopoiesis. Oncotarget. 2016.
Luo M, Jeong M, Sun D, et al. Long non-coding RNAs control hematopoietic stem cell function. Cell Stem Cell. 2015;16:426–38.
Tanos V, Ariel I, Prus D, et al. H19 and IGF2 gene expression in human normal, hyperplastic, and malignant endometrium. Int J Gynecol Cancer. 2004;14:521–5.
Yoshimizu T, Miroglio A, Ripoche MA, et al. The H19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci U S A. 2008;105:12417–22.
Gabory A, Jammes H, Dandolo L. The H19 locus: role of an imprinted non-coding RNA in growth and development. Bioessays. 2010;32:473–80.
Guo G, Kang Q, Chen Q, et al. High expression of long non-coding RNA H19 is required for efficient tumorigenesis induced by Bcr-Abl oncogene. FEBS Lett. 2014;588:1780–6.
Tsang WP, Ng EK, Ng SS, et al. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis. 2010;31:350–8.
Cai X, Cullen BR. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA. 2007;13:313–6.
Keniry A, Oxley D, Monnier P, et al. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell Biol. 2012;14:659–65.
Venkatraman A, He XC, Thorvaldsen JL, et al. Maternal imprinting at the H19-Igf2 locus maintains adult haematopoietic stem cell quiescence. Nature. 2013;500:345–9.
Bouchie A. First microRNA mimic enters clinic. Nat Biotechnol. 2013;31:577.
Ren S, Wang F, Shen J, et al. Long non-coding RNA metastasis associated in lung adenocarcinoma transcript 1 derived miniRNA as a novel plasma-based biomarker for diagnosing prostate cancer. Eur J Cancer. 2013;49:2949–59.
Crea F, Watahiki A, Quagliata L, et al. Identification of a long non-coding RNA as a novel biomarker and potential therapeutic target for metastatic prostate cancer. Oncotarget. 2014;5:764–74.
Wahlestedt C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat Rev Drug Discov. 2013;12:433–46.
Sun J, Song Y, Chen X, et al. Novel long non-coding RNA RP11-119 F7.4 as a potential biomarker for the development and progression of gastric cancer. Oncol Lett. 2015;10:115–20.
Zhou X, Yin C, Dang Y, et al. Identification of the long non-coding RNA H19 in plasma as a novel biomarker for diagnosis of gastric cancer. Sci Rep. 2015;5:11516.
Qiu JJ, Yan JB. Long non-coding RNA LINC01296 is a potential prognostic biomarker in patients with colorectal cancer. Tumour Biol. 2015;36:7175–83.
Tong X, Gu PC, Xu SZ, et al. Long non-coding RNA-DANCR in human circulating monocytes: a potential biomarker associated with postmenopausal osteoporosis. Biosci Biotechnol Biochem. 2015;79:732–7.
Tong YS, Wang XW, Zhou XL, et al. Identification of the long non-coding RNA POU3F3 in plasma as a novel biomarker for diagnosis of esophageal squamous cell carcinoma. Mol Cancer. 2015;14:3.
Meng L, Ward AJ, Chun S, et al. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409–12.
Yang Y, Cai Y, Wu G, et al. Plasma long non-coding RNA, CoroMarker, a novel biomarker for diagnosis of coronary artery disease. Clin Sci (Lond). 2015;129:675–85.
Velu CS, Chaubey A, Phelan JD, et al. Therapeutic antagonists of microRNAs deplete leukemia-initiating cell activity. J Clin Invest. 2014;124:222–36.
Dorrance AM, Neviani P, Ferenchak GJ, et al. Targeting leukemia stem cells in vivo with antagomiR-126 nanoparticles in acute myeloid leukemia. Leukemia. 2015;29:2143–53.
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–68.
Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494–501.
Papa V, Tazzari PL, Chiarini F, et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia. 2008;22:147–60.
Tazzari PL, Tabellini G, Ricci F, et al. Synergistic proapoptotic activity of recombinant TRAIL plus the Akt inhibitor Perifosine in acute myelogenous leukemia cells. Cancer Res. 2008;68:9394–403.
Konopleva MY, Walter RB, Faderl SH, et al. Preclinical and early clinical evaluation of the oral AKT inhibitor, MK-2206, for the treatment of acute myelogenous leukemia. Clin Cancer Res. 2014;20:2226–35.
Sandhofer N, Metzeler KH, Rothenberg M, et al. Dual PI3K/mTOR inhibition shows antileukemic activity in MLL-rearranged acute myeloid leukemia. Leukemia. 2015;29:828–38.
Park S, Chapuis N, Saint Marcoux F, et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia. 2013;27:1479–86.
Chiarini F, Lonetti A, Teti G, et al. A combination of temsirolimus, an allosteric mTOR inhibitor, with clofarabine as a new therapeutic option for patients with acute myeloid leukemia. Oncotarget. 2012;3:1615–28.
Amadori S, Stasi R, Martelli AM, et al. Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients with acute myeloid leukaemia: results of a phase II GIMEMA study (AML-1107). Br J Haematol. 2012;156:205–12.
Attar EC, Amrein PC, Fraser JW, et al. Phase I dose escalation study of bortezomib in combination with lenalidomide in patients with myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Leuk Res. 2013;37:1016–20.
Horton TM, Perentesis JP, Gamis AS, et al. A Phase 2 study of bortezomib combined with either idarubicin/cytarabine or cytarabine/etoposide in children with relapsed, refractory or secondary acute myeloid leukemia: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2014;61:1754–60.
Zeng Z, Shi YX, Samudio IJ, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6215–24.
Funding
This work was supported by National Basic Research Program (973) of China (2014CB541804, 2015CB910502) and Natural Science Foundation of China (91640101, 81472611).
Availability of data and material
Not applicable.
Authors’ contributions
XW wrote the manuscript. J-LC and SH revised and approved the final manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, X., Huang, S. & Chen, JL. Understanding of leukemic stem cells and their clinical implications. Mol Cancer 16, 2 (2017). https://doi.org/10.1186/s12943-016-0574-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-016-0574-7