
Abstract
Background
At time of diagnosis, less than 10% of patients with pancreatic adenocarcinomas (PDAC) are considered to be immediately operable (i.e. resectable). Considering their poor overall survival (OS), only tumours without vascular invasion (NCCN 2017) should be considered for resection, i.e. those for which resection with disease-free margins (R0) is theoretically possible in absence of presurgery treatment. With regard to high R1 rates and undetectable locoregional and/or metastatic spreading prior to surgery explain (at least in part) the observed 1-year relapse and mortality rates of 50 and 25%, respectively. Today, upfront surgery followed by adjuvant chemotherapy is the reference treatment in Europe. The main limitation of the adjuvant approach is the low rate of completion of the full therapeutic sequence. Indeed, only 47 to 60% patients received any adjuvant therapy after resection compared to more than 75% for neoadjuvant therapy. No previous prospective study has compared this approach to a neoadjuvant FOLFIRINOX or FOLFOX chemotherapy for resectable PDAC.
Methods
PANACHE01-PRODIGE48 is a prospective multicentre controlled randomized non comparative Phase II trial, evaluating the safety and efficacy of two regimens of neo-adjuvant chemotherapy (4 cycles of mFOLFIRINOX or FOLFOX) relative to the current reference treatment (surgery and then adjuvant chemotherapy) in patients with resectable PDAC. The main co-primary endpoints are OS rate at 12 months and the rate of patients undergoing the full therapeutic sequence.
Discussion
The “ideal” cancer treatment for resectable PDAC would have the following characteristics: administration to the highest possible proportion of patients, ability to identify fast-progressing patients (i.e. poor candidates for surgery), a low rate of R1 resections (through optimisation of local disease control), and an acceptable toxicity profile. The neoadjuvant approach may meet all these criteria. With respect to published data on the efficacy of FOLFOX and mFOLFIRINOX, these two regimens are potential candidates for neoadjuvant use in the aim to optimising oncological outcomes in resectable PDAC.
Trial registration
ClinicalTrials.gov, NCT02959879. Trial registration date: November 9, 2016.
Similar content being viewed by others

Background
Pancreatic duct adenocarcinoma (PDAC) is one of the most lethal forms of cancer, with 5-year overall survival (OS) rates of less than 5% for all stages [1]. It is the fourth-ranked cause of death by cancer and its incidence is steadily increasing in most western countries [2]. At diagnosis, 80 to 85% of patients present locally advanced or metastatic disease, only 5 to 22% of whom can be curatively treated by resection which explains the poor prognosis for PDAC in general [3, 4].
It is established that surgery is the only potentially curative treatment. However, even in resectable PDAC, the oncological results regarding OS and/or health-related quality of life (HRQoL)) need to be optimized. Indeed, in the absence of any adjuvant or neoadjuvant therapy, the long-term outcomes of surgically treated patients are still poor, with a 5-year OS rate of about 10% [5,6,7].
These very poor oncological results may be due to the presence of micrometastases or minimal residual disease not detectable at the time of surgery or spreading of cancer cells into the portal vein, lymphatic vessels and the peritoneal cavity following surgical manipulation of the tumour. Lymph node involvement (considered as a marker of early tumour dissemination) is found in more than 65% of cases [8]. According to the last American National Comprehensive Cancer Network classification (NCCN 2017) [9], a “immediately resectable” PDAC is defined by no tumor contact with the superior mesenteric vein (SMV) or portal vein (PV) or ≤ 180° contact without vein contour irregularity and no arterial contact.
However, a recent prospective analysis of PDAC resection specimens (using a standardized pathological reporting protocol) has demonstrated that even according to the “redefinition” of margin status, the frequency of positive resection margins (R1) is high (between 61 and 85%) [10,11,12]. R1 resection is a validated and robust prognostic factor after curative intent resection of PDAC [5,6,7, 13, 14].
Moreover, pancreatic surgery is associated with a high frequency of post-operative complications [15, 16], and chronic digestive and metabolic sequelae [17]. This is the main argument for optimizing patient selection and using the optimized surgical modality in good candidates in order to prevent palliative resection and to maintain HRQoL).
Adjuvant chemotherapy: Current standards of care
Randomized trials have suggested that both adjuvant 5-fluorouracil (5-FU) and gemcitabine improve median OS of 2.6–4.5 months and 2-year OS rate of 6 to 10%, when compared with pancreatic resection alone. No significant difference was demonstrated regarding either HRQoL and OS between these two drugs [5,6,7, 18, 19]. Gemcitabine is today’s drug of choice for adjuvant chemotherapy because better tolerated [7].
Recently, ESPAC 4 trial demonstrated that the combination of gemcitabine and capecitabine allowed a median overall survival of 28 months compared with 25.5 months following gemcitabine alone (p = 0.032) [14]. However, disease free survivals (DFS) were not statistically different between the two arms.
Long-term results are still poor, with 5-year OS rates of about 10 and 29% in the presence and absence of adjuvant chemotherapy, respectively [5, 6, 14, 19].
One limitation of the adjuvant approach was the low rate of completion of the full therapeutic sequence due to postoperative complications and poor performance status. Indeed, only 47 to 60% of patients received any adjuvant therapy after resection [20,21,22]. From an oncological point of view, completion of the full therapeutic sequence (surgery + adjuvant therapy) is well known to be strongly associated with favourable OS among patients with resectable PDAC [22, 23].
In the subset of patients receiving adjuvant treatment, pancreatic cancer exhibits a prominent tendency to recur locally and to metastasize after a short period of time; 1-year OS and DFS rates are 75 and 50%, respectively [5, 6, 24]. The pattern of disease failure corresponds to extrapancreatic dissemination in 63 to 83% of patients and isolated local failure for 17 to 37% of patients [5, 7, 18].
Oncological treatement accessibility
Despite the theoretical benefits discussed above, there is currently no unambiguous evidence in favour of routine clinical use of neoadjuvant therapy in resectable PDAC. In fact, several single-arm studies have reported heterogeneous results, with median OS and 2-year OS rates ranging from 15 to 35 months and 27 to 55%, respectively [25,26,27,28,29,30,31]. These results cannot be compared directly, due to differences in trial design. Nevertheless, the published long-term results are better than those achieved in modern series of patients undergoing surgery alone [5, 24] (median OS: 18–20 months; 2-year OS rate: 40–42%) and appear to be of the same order of magnitude or moderately better than those observed in series of patients treated with adjuvant single-agent chemotherapy [5, 7, 18, 24] (median survival: 21–25 months, 2-year OS rate: 41–48%) or combination chemotherapy [32,33,34] (median survival: 25.4–32.1 months; 2-year OS rate: 59–80%). A systematic review and meta-analysis of 111 trials confirmed that neoadjuvant treatment does not seem to provide any benefit over adjuvant therapy in resectable PDAC [20].
However, inter-trial comparisons (which already have many serious limitations) are further hampered in this particular case by the fact that the typical population enrolled in adjuvant trials is better selected than those in neoadjuvant trials. For example, 15% of the tumours staged as resectable cannot be resected due to undetected metastatic disease or underestimated vascular involvement, furthermore up to 30% of the patients are not suitable to receive adjuvant therapy because of poor postoperative performance status. Both groups of patients cannot be enrolled into adjuvant trials, thus improving OS by simple patient selection.
An additional argument to support neoadjuvant treatment has been recently published in a report of the extracted data of the American National Cancer Database [35]. Indeed, Mokdad et al. demonstrated that neoadjuvant treatment followed by resection was associated with a significant OS benefit compared to upfront resection in early-stage resected PDAC, with a median survival of 26 months and 21 months, respectively (p = 0.01).
FOLFIRINOX as a potential candidate to improve oncological results
Since the first usage of FOLFIRINOX in the setting of advanced PDAC by Conroy et al. in 2005, and the result of the randomized control trial of the PRODIGE intergroup in metastatic PDAC, this polychemotherapeutic regimen has been used widely [36,37,38,39]. Following FOLFIRINOX based treatment, the overall R0 resection rates were about 70–77% [37] and 84–92% [38, 39], respectively for locally advanced and borderline PDAC. In addition, pathologic complete response could be achieved in 7 to 13% in cases.
FOLFOX as a potential alternative to FOLFIRINOX
Recently, the NCCN stated that Fluoropyrimidines plus oxaliplatin combination therapy can be an acceptable option for patients with gemcitabine refractory PDAC [9].
In a prospective phase II trial that sought to evaluate sequential FOLFOX-6 and gemcitabine followed by appropriate maintenance treatment for advanced pancreatic cancer, the clinical response rate was 44% with an acceptable and manageable toxicity profile [40]. Morever, in patients with gemcitabine refractory PDAC, the combination of oxaliplatin and 5-FU was superior to 5-FU alone or best supportive care in the CONKO-003 trial [41].
Progress is needed, novel drugs have to be tested and alternative strategies evaluated - mainly because adjuvant therapy may not provide any benefit to between a third and a half of patients regarding surgical complications and does not have any impact on the resection margin - one of the main prognostic factors for OS.
Methods/design
Study design
PANACHE-PRODIGE-48 is a prospective open, non-comparative, randomized, multicentre phase II study designed to assess the safety and efficacy of two modes regimens of neoadjuvant chemotherapy (mFOLFIRINOX & FOLFOX) relative to the current reference treatment (surgery follow by adjuvant chemotherapy) for resectable PDAC.
Patients with resectable PDAC (definition based on the NCCN’s) [9] will be randomised to either pancreatectomy and adjuvant chemotherapy or 4 cycles of neoadjuvant chemotherapy with either FOLFOX or mFOLFIRINOX. The patients in the neoadjuvant chemotherapy arms will receive postoperative chemotherapy for 4 months (allowing a 6 months total duration of chemotherapy).
For each patient, treatment time is approximately 7–8 months, with an anticipated maximum period of 9 months.
The primary analysis will be performed on a co-primary endpoint overall survival rate at 12 months after randomisation and the rate of patients undergoing the full therapeutic sequence. An intermediate analysis will be performed once 27 patients evaluable for the co primary endpoint with 12 months of follow-up have been included in each of the 2 arms. The study is planned for a total duration of 5 years. The secondary analyses will be performed 3 years after the last inclusion.
The study is registred on clinicaltrial.gov website (NCT02959879).
Primary objective
The primary objective of the trial is to evaluate the feasibility and efficacy of two regimens of neoadjuvant chemotherapy. The co-primary endpoints are the observed OS rate at 12 months and the rate of patients undergoing the full therapeutic sequence.
OS is defined as time from randomisation to death from any cause. Alive patients will be censored at the last follow-up [42]
Feasibility is defined as the rate of patients having undergone the full therapeutic sequence (regardless of the study arm). Specific definitions are required in each study arm and so “full therapeutic sequence” are defined as:
-
“two or more cycles of neo-adjuvant chemotherapy followed by surgical resection” in the neoadjuvant arms.
-
“surgical resection completed by four or more cycles of adjuvant chemotherapy” in the control arm.
Hence, this parameter represents an objective evaluation in the control arm (no neoadjuvant chemotherapy) and the two neoadjuvant chemotherapy arms (mFOLFIRINOX & FOLFOX).
Secondary endpoints
-
One and 3-year DFS (DATECAN consensus definition for the pancreas – DFS is defined by time after treatment during which no disease is found with respect to study follow-up) [42, 43]
-
3-year OS. OS is defined as the interval between the randomization date and the date of death from any cause.
-
HrQoL (EORTC QLQ C-30 and QLQ-PAN26 questionnaires) The QLQ-PAN26 module is a specific module for pancreatic cancer, currently in phase IV validation by EORTC [44].
-
Chemotherapy-related toxicity, graded according to CTCAE V4.0.
-
Overall postsurgical morbidity, graded according to Dindo/Clavien classification [45],
-
Specific morbidity due to pancreatic fistula, graded according to the International Study Group of Pancreatic Fistula (ISGPF) criteria [46].
-
Histological tumor response (according to the College of American Pathologists (CAP) grading system)
-
Radiological tumour response (according to RECIST v1.1) will be studied. Radiological data will be stored on a dedicated platform.
-
Constitution of tissue and serum banks.
Eligibility criteria
-
Inclusion criteria
For inclusion in the study, all of the following inclusion criteria must be fulfilled:
-
○ Signed and dated informed consent
-
○ Patients willing and able to comply with protocol requirements.
-
○ Histology-proven, adenocarcinoma of the pancreas.
-
○ Resectable adenocarcinoma (according to NCCN classification 2017) [9]: the absence of distant organ or distal lymph node metastases, the absence of evidence of SMV and PV distortion, tumour thrombus, or venous encasement, the existence of clear fat planes around the celiac axis, hepatic artery and superior mesenteric artery (SMA). Resecability is evaluated on arterial-phase and portal-phase IV contrast-enhanced multislice CT-scan of the pancreas (slice thickness: 2.5 mm) and Diffusion Weighted MRI of liver, as evaluated in a multidisciplinary staff meeting including at least one radiologist and one expert surgeon.
-
○ No prior chemotherapy.
-
○ Age ≥ 18 or < 85 years
-
○ ECOG performance status 0–1.
-
○ Adequate hematologic function: neutrophils > 1.5 × 109/L; platelets > 100 × 109/L; hemoglobin ≥10 g/dL (transfusions are authorized).
-
○ Adequate renal function: creatinine clearance (according to Cockroft and Gault’s equation) > 60 ml/min
-
○ Adequate liver function: AST (SGOT) and ALT (SGPT) ≤ 2.5 x ULN (≤5 x ULN in case of liver metastases), total bilirubin ≤1.5 x ULN.
-
○ Baseline evaluations performed before randomization: clinical and blood evaluations no more than 14 days prior to randomization, tumor assessment (thorax-abdominal-pelvis CT-scan and liver MRI) no more than 21 days prior to randomization.
-
○ Women of child-bearing age not having undergone a hysterectomy or tubal ligation must undergo a negative pregnancy test (i.e. normal serum or urine beta-HCG level) before inclusion, and should use effective contraception throughout the study and for the following 6 months.
-
Exclusion criteria
Patients are not eligible for this study if any of the following criteria apply:
-
○ PDAC defined as “borderline”, locally advanced non-resectable or metastatic.
-
○ Prior cancer therapy for PDAC
-
○ Surgical or anaesthesiological contra-indications:
-
○ non-controlled congestive heart failure - non-treated angina – recent myocardial infarction (in the previous year) – non-controlled arterial hypertension (SBP > 160 mm or DBP > 100 mm, despite optimal drug treatment), long QT.
-
○ major non-controlled infection.
-
-
○ Any medical, psychological or social situation that (in the investigator’s opinion) could limit (i) the patient’s compliance with the protocol or (ii) the ability to obtain or interpret data.
-
○ History or current evidence on physical examination of central nervous system disease or peripheral neuropathy ≥ grade 1, according to according to Common Terminology Criteria for Adverse Events (CTCAE) v.4.0.
-
○ Known hypersensitivity reaction to any of the components of study treatments.
-
○ Pregnancy (the absence of which must be confirmed in a ß-hCG test) or breast-feeding.
-
○ Any significant disease which, in the investigator’s opinion, would exclude the patient from the study.
-
○ Patients having been included in a clinical trial within the previous 4 weeks or participating in another trial.
Randomization
After completion of all the screening evaluations (compliance with all the inclusion criteria and none of the exclusion criteria) and signature of the informed consent forms, all eligible patients will be randomly assigned (2:2:1) to the three arms using minimisation technique, as follows:
-
The “mFOLFIRINOX” arm: neo-adjuvant chemotherapy – surgical resection– adjuvant chemotherapy
-
The “FOLFOX” arm: neo-adjuvant chemotherapy – surgical resection– adjuvant chemotherapy
-
The “Standard” arm: immediate surgical resection, followed by adjuvant chemotherapy (according to current guidelines)
Centralized randomization using minimisation technique will be stratified according to the study centre, the topography of tumor (uncinate/head/neck versus body/tail), bilirubin level (< 1.5 N vs. > 1.5 N) and the CA19–9 level (≤200 U/ml vs. > 200 U/ml).
Enrolment
One hundred sixty evaluable patients need to be enrolled and randomized in a 2:2:1 ratio, with 64 patients in both the mFOLFIRINOX and FOLFOX arms, and 32 patients into the control arm.
Assuming a 5% dropout rate a total of 168 patients need to be recruited to attain the necessary power for the statistical analyses.
The accrual duration will be of 24 months.
Ethics
This study is conducted in accordance to the standards of Good Clinical Practice (ICH-E6), the European Directive 2001/20/EC, the revised version of the Declaration of Helsinki, and local regulations. The protocol has been submitted and approved by the Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM; French National Agency for Medicines and Health Product Safety) and the Comité de Protection des Personnes – NORD OUEST I (French Ethics Committee). Written informed consent is obtained from all patients prior to randomization.
Pre-therapeutic workup
Prior to inclusion in the trial an information booklet will be given to each patient and the trial protocol will be clearly explained. The initial work-up must take place in the 4 weeks preceding randomization, and the maximum time between starting procedures and randomisation will be 21 days.
The screening assessments (this initial work-up) will comprise:
-
A complete physical examination
-
Biological assessments. Estimation of full blood count, calculation of creatinine clearance using the Cockroft formula, measurements of electrolytes, total protein, albumin, alkaline phosphatase, aspartate transaminase, alkaline transaminase, total and conjugated bilirubin, gamma-GT, pregnancy test for females of reproductive years and discussion of contraceptive methods during the study period
-
Tumor markers evaluation: Carbohydrate antigen 19–9
-
Assessment of operability – Electrocardiogram, cardiac echography, anaesthetic consultation.
-
CT scan: Patients eligible for this study have resectable PDAC as shown on CT scan of thorax, abdomen and pelvis with IV contrast. As recommended by the 2015 NCCN Clinical Practice Guidelines in Oncology, the CT scan should be performed according to a defined pancreas protocol (such as three-phase cross-sectional imaging with thin slices). This technique allows accurate visualisation of the relationship between the primary tumour and the mesenteric vasculature. To this end, we shall use an optimal multiphase imaging technique that includes a non-contrast phase plus arterial, pancreatic parenchymal and portal venous phases of contrast enhancement with thin slices (2.5 mm) through the abdomen. The Pre-operative CT-scan should have been done during the 30 days period before the exact date of surgery. We shall use the latest NCCN radiological criteria (NCCN 2017) [9].
-
Diagnostic confirmation by echoendoscopy with fine needle aspiration. Histopathologic confirmation of PDAC diagnosis is required before randomization. Systematic endoscopic ultrasound with fine needle aspiration or fine needel biopsy will be performed. The technique will be repeated in the event of initial negative results.
-
Liver Diffusion Weighted MRI: MRI is mandatory for the detection of hepatic lesions undiagnosed by CT. By adding hepatic MRI during preoperative evaluation, hepatic metastases were newly discovered in 5% patients (5%) without hepatic lesions on CT scan [47, 48].
-
Staging laparoscopy is recommended, especially if certain unfavorable prognostic factors are highlighted (T ≥ 3 cm CA 19–9 ≥ 200 U/ml tumor body or tail of the pancreas, back pain; greater than 10% weight loss).
During this visit, inclusion and exclusion criteria will be checked and validated. Each part of the screening evaluation will be reported on a specific form (endoscopic, clinical, biological and radiological data). Informed consent forms for the clinical study and translational research (blood samples) will be handed out to the patient. The following assessments and procedures will be performed and documented:
Treatment methods and application
After completion of all the screening evaluations (compliance with all the inclusion criteria and none of the exclusion criteria) and signature of the informed consent forms, all eligible patients will be randomly assigned (2:2:1) to the three arms, as follows:
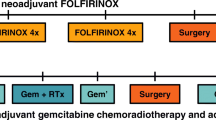
ARM 1 Neoadjuvant mFOLFIRINOX: neo-adjuvant chemotherapy – surgical resection– adjuvant chemotherapy
-
Step 1: Neoadjuvant mFOLFIRINOX
Must start within 21 days of randomization, if bilirubin level is below 1.5 N.
Biliary drainage will be performed before the first cycle of chemotherapy administration, if bilirubin level is above 1.5 N in the mFOLFIRINOX arm. If drainage fails despite an additional endoscopic procedure, the neoadjuvant sequence will be abandoned.
4 cycles of mFOLFIRINOX neo-adjuvant chemotherapy.
Clinical and biochemical assessments (CA 19–9 assay and collection of a serum samples will be performed after the two first cycles and within 28 days of the end of the 4th cycle.
Radiological assessment (CT scan of the thorax, abdomen and pelvis) within 28 days of the end of the 4th cycle.
-
Step 2: Surgery
Performed 3 to 5 weeks after the end of the last cycle of neoadjuvant chemotherapy.
Pancreatic tumour resection according to current French guidelines
-
Step 3: Adjuvant chemotherapy
Adjuvant chemotherapy for 4 months; the choice of drug or combination of drugs will be left to the medical teams, according to current guidelines and practice validated during the recruitment period.
ARM 2 Neoadjuvant FOLFOX: neo-adjuvant chemotherapy – surgical resection– adjuvant chemotherapy
-
Step 1: Neoadjuvant FOLFOX
Must start within 21 days of randomization, if bilirubin level is below 1.5 N.
Even though biliary drainage is not mandatory in the FOLFOX arm, it is advisable and the decision will be left to the attending physicians.
4 cycles of FOLFIRINOX neo-adjuvant chemotherapy.
Clinical and biochemical assessments (CA 19–9 assay and collection of a serum samples will be performed after the two first cycles and within 28 days of the end of the 4th cycle.
Radiological assessment (CT scan of the thorax, abdomen and pelvis) within 28 days of the end of the 4th cycle.
-
Step 2: Surgery
Performed 3 to 5 weeks after the end of the last cycle of neoadjuvant chemotherapy.
Pancreatic tumour resection according to current French guidelines
-
Step 3: Adjuvant chemotherapy
Adjuvant chemotherapy for 4 months; the choice of drug or combination of drugs will be left to the medical teams, according to current guidelines and practice validated during the recruitment period.
ARM 3 UPFRONT SURGERY: Immediate surgical resection, followed by adjuvant chemotherapy (according to current French guidelines)
-
Step 1: Surgery
Performed within 21 days of randomization.
Pancreatic tumour resection according to current French guidelines
-
Step 2: Adjuvant chemotherapy
Adjuvant chemotherapy for 6 months; the choice of drug or combination of drugs will be left to the medical teams, according to current guidelines and practice validated during the recruitment period.
The therapeutic scheme for this trial is depicted in Fig. 1.

Protocol overview. MRI: Magnetic resonance imaging; PDAC: Pancreatic duct adenocarcinoma; TDM TAP: CT-scan Thorax Abdomen and Pelvis
Key point of biliary drainage
Patients with bilirubin level greater than 250 micromol/l or cholangitis at presentation required systematic biliary drainage, based on the consensual indications of pre-operative biliary drainage before any surgical procedure or chemotherapy [49]. It is well accepted that the optimal duration of biliary drainage is 4 to 6 weeks. Endoscopic pre-operative biliary drainage during endoscopic retrograde cholangiopancreatography with coated self-expanded metal stents is recommended [50]. Percutaneous transhepatic biliary drainage is an option after failure of endoscopic procedure.
Otherwise, eligible patients will be randomized to either surgery alone or neoadjuvant chemotherapy.
If bilirubin level is greater than 1.5 N in patients randomized to the FOLFIRINOX arm, drainage will be implemented in all cases.
Even though biliary drainage is not mandatory in the FOLFOX arm, it is advisable and the decision will be left to the attending physicians.
Chemotherapy regimens
Side-effects of chemotherapy are graded by the “Common Terminology Criteria for Adverse Events” version 4 https://evs.nci.nih.gov/ftp1/CTCAE/About.html
At each study visit, laboratory parameters are determined for dose adjustments.
-
Neoadjuvant (pre-operative chemotherapy)
-
○ mFOLFIRINOX Group
-
The modified FOLFIRINOX regimen (mFOLFIRINOX) will be administered intravenously over 48 h once every 2 weeks, for 2 months (i.e. on D1/D2, D15/D16, D29/D30 and D43/D44).
The mFOLFIRINOX regimen will be administered as follows: oxaliplatin will be administered as an 85 mg/m2 intravenous infusion over 2 h (on D1 only) concomitantly with irinotecan (as an 180 mg/m2 intravenous infusion over 90 min (on D1 only)) and LV (as a 400 mg/m2 infusion over 2 h), followed by 5-FU (given as a 2400 mg/m2 continuous infusion over 48 h, without previous bolus injection).
The cycle length is 2 weeks, comprising 48 h of infusion and 12 days of rest. These cycles must be repeated every second week. A total of 4 preoperative cycles will be administrated to all patients in the mFOLFIRINOX arm.
-
○ FOLFOX Group
The FOLFOX regimen will be administered intravenously over 48 h once every 2 weeks, for 2 months (i.e. on D1/D2, D15/D16, D29/D30 and D43/D44).
The FOLFOX regimen will be administered as follows: oxaliplatin will be administered as an 85 mg/m2 intravenous infusion over 2 h (on D1 only) concomitantly with LV (as a 400 mg/m2 infusion over 2 h), followed by 5-FU (given as a 400 mg/m2 bolus injection over 10 min, and then as a 2400 mg/m2 continuous infusion over 46 h).
The cycle length is 2 weeks, which comprises 48 h of infusion and 12 rest days. These cycles must be repeated every second week. A total of 4 preoperative cycles will be administered to all patients in the FOLFOX arm.
-
○ Re-staging post neoadjuvant chemotherapy
Tumour restaging will be performed within 28 days of the end of the 4th cycle of neoadjuvant chemotherapy. It will comprise: clinical examination, same blood analyses as at inclusion, tumour marker estimation (CA19.9), anaesthetic consultation, CT-scan of the thorax, abdomen and pelvis, and liver Diffusion Weighted MRI. This re-evaluation will be performed by the same team and by the same technique as the initial work-up.
Surgical resection
The choice of the surgical approach will be left to the medical teams according to their experience in laparoscopic pancreatic surgery. Surgery must be performed 3 to 5 weeks after the end of the last cycle of neoadjuvant chemotherapy or as soon as possible in the control arm.
Surgical resection will follow the French national guidelines for GA, published by the SFCD-ACHBT-HAS-INCA [51].
-
Pancreaticoduodenectomy
During pancreaticoduodenectomy, a para-aortic lymph node sampling is required, and should be evaluated using frozen-section analysis. Circumferential dissection of the portal vein–superior mesenteric vein (PV-SMV) axis and dissection of the right hemicircumference of the superior mesenteric artery (SMA) to the right of the coeliac trunk were required. The surgeon clearly identified the margins in the operative room with multicolour coded inking of: (i) the mesenterico–portal vein groove or PV-SMV margin (PV-SMVm); (ii) the SMA margin (SMAm), and (iii) the posterior margin.
“En-bloc” standard lymphadenectomy will be performed systematically including peripancreatic nodes (groups 13 and 17), nodes along the hepatic artery (group 8), hepatoduodenal ligament nodes (group 12), supra- and infrapyloric nodes (groups 5 and 6) and periadventitial dissection of the right lateral aspect of the SMA (groups 14b and 14c).
-
Distal pancreatectomy
In the treatment of left-sided pancreatic cancer, radical antegrade modular pancreatosplenectomy (RAMPS) should be preferred [52] .
Resection of lymph nodes along the splenic artery (group 11), at the splenic hilum (group 10), and along the inferior margin of the pancreas (group 18) is recommended during distal pancreatectomy.
Pathological analysis
A standard nomenclature should be used to describe surgical margins. For pancreatoduodenectomy specimens, the status of the pancreatic neck margin, bile duct margin, anterior surface, posterior surface, portal vein/SMV groove, and SMA/uncinate margin should be described. For distal pancreatectomy specimens, the anterior surface, posterior surface, and pancreatic transection margins should be described. The pathological protocol also included the maximal transverse diameter of the tumour, the distance between the tumor and the different surgical margins (mm) the tumour–node–metastasis (TNM) classification, the grade of differentiation, the presence or absence of perineural, lymphatic and/or vascular spread, the number of lymph nodes retrieved from the specimen, enabling the calculation of the lymph node ratio (LNR) and the pathological tumor response rate (according to the CAP grading system).
Serial slicing of the entire pancreatic specimen will be performed according to the guidelines of the Royal College of Pathologists and the Leeds Pathology Protocol [12, 53]. Margin involvement (R1) is defined for the 0-mm margin if tumour cells were present at the inked margin; R1 is also defined for each margin width if tumour cells were present within the margin, independently of the mode of tumour spread; The resection was considered as curative (R0) if no tumour cells were identified at any of the resection margins, again for each margin width.
Patient follow-up
After surgery, participating patients will be reviewed in out-patient consultations 1 month (+/− 14 days) after hospital discharge. They will have a full clinical examination, biochemical analysis, tumour marker estimation (CA19.9), thorax-abdominal-pelvis CT scan. Questionnaires will be completed with regards to quality of life (QoL), emotional state, emotional adjustment, perception of illness and lifestyle burden.
After completion of adjuvant chemotherapy, a further consultation will be scheduled for one month (+/− 14 days) with evaluation of clinical state, treatment tolerance, haematological parameters, renal function, tumour markers, and the same set of questionnaires will be completed.
All participants will subsequently be followed for 3 years after randomization. Out-patient consultations will be scheduled every 3 months (+/− 15 days) and patients will be evaluated by clinical examination, thoraco-abdominal-pelvic CT scan, tumour marker estimation (CA19.9) and QoL questionnaires.
Further examinations will be requested as required.
If disease recurrence is suspected its site (loco regional or distant) must be documented by the appropriate investigations and histological proof be obtained wherever possible. In cases of strong clinical suspicion of recurrence without radiological or histological proof, surgical exploration is recommended to identify peritoneal carcinomatosis not visualised by standard radiological investigation. All cases of disease recurrence will be discussed at the multi-disciplinary team meeting and further therapeutic strategies recorded.
Reason for discontinuing treatment protocol
The treatment protocol may be discontinued for the following reasons: decision of the treating doctor, high grade toxicity, occurrence of a serious adverse effect, disease progression, patient refusal to continue and patient death. In cases where treatment is discontinued, patients will remain within the intention to treat analysis.
Samples size calculation and statistical considérations
According to Bryant and Day two-stage design [54] with a ration of 2:2:1, it will be required to randomize 64 patients into the FOLFOX arm, and 64 patients into the FOLFIRINOX arm (with a one-sided type 1 error of 5% and a power of 85%) to verify the following hypotheses:
-
Hypothesis H0: an observed OS rate at 12 months of 70% and a feasibility of the full therapeutic sequence in 55% of patients in the neo-adjuvant FOLFOX and FOLFIRINOX groups will be considered as uninteresting to pursue evaluation in Phase III
-
Hypothesis H1: The expected outcome corresponds to an observed OS rate at 12 months of 85% and a feasibility in 75% of patients in the neo-adjuvant FOLFOX and FOLFIRINOX groups
In first stage, an intermediate analysis will be performed once 27 evaluable patients with 12 months of follow-up have been included in each of the 2 arms.
-
Upper limit for rejecting drug interest due to inadequate OS rate at 12 months will be 19 deaths
-
Upper limit for rejecting drug interest due to inadequate feasibility toxicity will be 15 patients without full therapeutic sequence
In other case we will pursue the inclusion by including 37 additional patients in each arm reaching the criteria for a total of 64 evaluable patients
-
Upper limit for rejecting drug interest due to inadequate OS rate at 12 months will be 50 deaths
-
Upper limit for rejecting drug interest due to inadequate feasibility toxicity will be 41 patients without full therapeutic sequence
In other case treatment will considered as promising and will be regarded as interesting for further evaluation in a phase III trial.
Taking into account 32 additional patients to be randomized in the control arm to validate our hypotheses (especially H0) due to the 2:2:1 randomised ratio, 160 patients should be randomized.
Taking in account, 5% of patients lost to follow up 168 patients will need to be enrolled.
In case of positive results of the phase 2 we plan to continue with a phase III trial at the end of the phase II. The phase 2 will then constitute an interim analysis of phase III (O Brien Fleming boundaries with alpha spending function) in this case statistical comparison will be done in the line of phase III design with stringent marging to reject earlier H0 or H1 (futility), such results would not be communicated in case of no rejection. Patients randomized and included in the phase II will be kept for phase III according to phase II results.
A statistical analysis plan will be written before data frozen. All changes to this plan will be documented. Since this a non comparative randomized phase II trial, statistical comparisons will be done in a exploratory purpose.
Discussion
The present trial offers the unique opportunity to explore the interest of 2 neoadjuvant chemotherapy regimens (FOLFOX, mFOLFIRINOX) in contrast of immediate surgery with adjuvant chemotherapy in the setting of resectable PDAC.
Neoadjuvant strategy in resectable PDAC
In theory, neoadjuvant treatment has several potential advantages in the context of resectable PDAC.
In terms of the current standard management of surgery with adjuvant therapy, between 40 and 50% of patients having undergone curative resection do not receive the adjuvant treatment planned due to surgical complications, poor performance status, comorbidity, patient refusal, and/or early disease recurrence [21,22,23, 55,56,57,58]. Otherwise, completion of the full therapeutic sequence after neoadjuvant treatment was observed in 75% in a recent report from an experienced centre [21]. Hence, neoadjuvant strategy may allow administrating the full therapeutic sequence to a greater proportion of patients than adjuvant strategy alone.
Neoadjuvant therapy may achieve down-staging, which in turn improves the R0 resection rate and increases the locoregional control. Indeed, Schorn reported that after neoadjuvant therapy, the rates of vascular and perineural involvement were lower than for immediately resected tumours [59]. Moreover, significantly lower positive lymph node (LN) rates were observed after neoadjuvant treatment (between 10 and 40%) [27, 30, 59] whereas lymph node involvement is constant in case of upfront surgery - between 65 and 80% of cases [60, 61].
Although complete resection of these “resectable” tumours would seem to be technically possible, the actual R1 resection rate is liable to be high. As mentioned above evoked, a recent prospective analysis of pancreatic resection specimens (using a standardized pathological reporting protocol) has demonstrated that according to the “redefinition” of margin status, the frequency of positive-margin resection (R1) is as high as 70%. The R1 resection rate was 60% in the ESPAC-4 trial [14]. This value provides a rationale for neoadjuvant treatment, with the hope of improving the rate of R0 resection.
Indeed, recent studies on neoadjuvant therapy in a context of borderline resectable disease, confirmed this hypothesis, with regard to achievement of R0 resection in more than 90% of cases [34, 36, 62].
The concern for disease progression during neoadjuvant treatment is not negligible, due to the low rate of curative resections performed in patients whose disease was deemed resectable at the time of starting neoadjuvant therapy - both after induction chemoradiation (45–74%) [25, 30, 31] and after induction chemotherapy (38–70%) [28, 63]. However, advocates of neoadjuvant therapy claim that these data show the value of this strategy and argue that patients who experience disease progression during induction treatment are suffering from an extremely aggressive tumour and undiagnosed initial micrometastatic disease that cannot be cured by extensive surgery. This type of strategy may help select good candidates for surgery. It would be desirable to reduce or avoid the risk of surgical mortality and morbidity in this subset of patients.
Which neoadjuvant chemotherapy?
Even though single-agent gemcitabine and 5FU have been validated in adjuvant and metastatic settings, the objective response was low, at around 10% [64, 65], whereas combination chemotherapy yielded a response rate of 9–28% in advanced PDAC [66,67,68,69,70]. Interestingly, using more than two chemotherapeutic agents in advanced PDAC increased the response rate: the three-drug combinations were gemcitabine, a fluoropyrimidin and a platinating agent (18–41%) [71,72,73,74], either FOLFIRINOX (5-FU-oxaliplatin-irinotecan; 27–50%) [75,76,77,78] or G-FLIP (gemcitabine-5FU-irinotecan-cisplatin; 26%) [79] and showed promising results.
The superiority of intensive polychemotherapy regimens was also suggested by an Italian survey of treatment trends and outcomes in large series of patients with stage III/IV PDAC [80]. Gemcitabine alone appeared to be significantly inferior to four-drug combinations, with median OS times of about 5.1 and 9.1 months, respectively. Based on these data and considerations, polychemotherapy regimens are suitable candidates in a neoadjuvant setting.
Moreover, even though gemcitabine is still one of the main weapons in the oncologist’s arsenal, recent studies have shown that two thirds of patients with PDAC do not benefit from gemcitabine based-chemotherapy in an adjuvant setting because of low or moderate expression levels of gemcitabine nucleoside transporters (hENT1) [81,82,83]. In this context, it will be interesting to more accurately define the subgroups of patients who would derive particular benefit from gemcitabine and those who should be treated with non-gemcitabine-based combinations. A pre-operative assay of hENT1 expression in biopsy samples is not currently feasible in multicentre trial even if studies are ongoing [84]. Hence, from an ethical point of view, we decided to compare two non-gemcitabine-based combinations: mFOLFIRINOX and FOLFOX.
mFOLFIRINOX
In 2005, Conroy et al. [75] evaluated the response rate and toxicity of FOLFIRINOX in an advanced PDAC setting. The study yielded promising results, with a response rate of 27%, a time to progression of 8.2 months, and an OS of 10.2 months. Despite the fact that grade 3/4 neutropenia occurred in 52% of patients, patients had improvement in all functional scales of the EORTC QLQ-C30 assessment questionnaire. Based on these data, Conroy and colleagues conducted a phase III trial comparing FOLFIRINOX with gemcitabine as first-line treatment for metastatic PDAC in 342 patients with good performance status (0–1) [76]. Median OS was 11.1 months in the FOLFIRINOX group and 6.8 months in the gemcitabine group (hazard ratio (HR): 0.57; 95% confidence interval (CI): 0.45–0.73; p < 0.001). Median progression-free survival (PFS) was 6.4 months vs. 3.3 months (HR: 0.47; 95% CI: 0.37–0.59, p < 0.001). The overall response rate was 31.6% vs. 9.4% (p < 0.001). Grade 3/4 toxicities were more common in the FOLFIRINOX group than in the gemcitabine group (diarrhoea 12.7% vs. 1.8%, nausea 15.6% vs. 6.3%, vomiting 14.5% vs. 8.3%, fatigue 23.6% vs. 17.8%, neutropenia 45.7% vs. 21%, and febrile neutropenia 5.4% vs. 1.2%, respectively). In the FOLFIRINOX arm, 42% of patients received support with granulocyte colony-stimulating factor.
One of the main criticisms of this trial was that only 39% of patients had a primary tumour in the head of the pancreas (possibly requiring biliary stents), whereas the proportion is about two-thirds in clinical practice. This type of polychemotherapy can cause severe neutropenia, which is a major concern in this setting because of the specific risk of cholangitis in patients with biliary stents. However, recent data suggest that after effective biliary drainage, FOLFIRINOX could be used even for locally advanced PDAC patients with jaundice, and is currently proposed as first line therapy for Borderline and Locally advanced PDAC [9, 85]. In the studies by Marthey et al. [78] and Hosein et al. [86], respectively 47 and 50% of patients required biliary drainage. No patient was taken off FOLFIRINOX for toxicity reasons, even though 26 and 40% respectively experienced grade 3/4 adverse events. This toxicity profile was managed by use of a granulocyte colony-stimulating factor analogue in up to 85% of cases and a FOLFIRINOX dose reduction in 20% of cases in both studies [78, 86]. The toxicity profiles did not appear to depend on the presence or absence of a stent. There was no episode of cholangitis during FOLFIRINOX treatment in any of the patients. Other recently published retrospective studies have shown similar results [77, 87], confirming that FOLFIRINOX could be a viable option even in patients with a biliary stent. Therefore, criteria such as good performance status, controlled bilirubin level and good supportive care must be complied with.
Given the toxicity of FOLFIRINOX, there is a pressing need for active, safe agents - especially for patients with poor performance status and who cannot risk exposure to the high toxicity of a combined regimen.
Modified FOLFIRINOX was better tolerated and as efficient as “classic” FOLFIRINOX in previous studies [84]. Moreover, the tolerability and feasibility of mFOLFIRINOX has been demonstrated in a phase I-II trial in patients with borderline PDAC [63].
FOLFOX
The oxaliplatin–5-FU-based combination also appears to be a valuable option. One of the oxaliplatin/5FU combinations, the FOLFOX 6 regimen, has shown a good response rate in pancreatic cancer [88, 89], with tolerable rates of grade 3/4 toxicity. In 2012, n et al. confirmed their promising results for the oxaliplatin/5FU combination regimen in a prospective phase II trial that sought to evaluate sequential FOLFOX-6 and gemcitabine followed by appropriate maintenance treatment for advanced pancreatic cancer. The clinical response rate and biochemical response rate (based on significant decrease in CA 19–9 levels) were 44% (22% for both the partial response and stable disease) and 64.3%, respectively. The toxicity profile of the FOLFOX-6 combination was acceptable and manageable [40].
Recently, the NCCN stated that Fluoropyrimidines plus oxaliplatin combination therapy can be an acceptable option for patients with gemcitabine refractory pancreatic cancer [9]. Several studies have reported infused 5-FU plus oxaliplatin as a second line treatment with a combined response and disease control rate of 17 to 52%, PFS of 1.4 to 4.0 months and OS of 3.4 to 6.7 months [90,91,92,93,94].
The main objective of our Phase II PANACHE-01 study is to assess the safety and efficacy of neo-adjuvant mFOLFIRINOX and FOLFOX in patients with resectable PDAC, relative to the current reference treatment (surgery and then adjuvant chemotherapy). The control arm will consist of patients treated with upfront surgery followed by adjuvant chemotherapy. This study will enable us to investigate the efficacy and tolerability of a neo-adjuvant treatment protocol and to select the best regimen for improving rates of long-term DFS and complete/sustained remission in these patients.
The most innovative aspect of this study would be its validation of the neoadjuvant approach as treatment for resectable PDAC, with the use of aggressive drugs (FOLF(IRIN)OX). To date neoadjuvant FOLF(IRIN)OX treatment has not been tested for resectable tumours.
In view of the limitations of the reference treatment (surgical resection followed by adjuvant chemotherapy) in terms of post-operative feasibility (fewer than 60% of patients can be treated) and lack of efficacy (in term of resection margins, node involvement, DFS and OS), other approaches must be considered.
The “ideal” cancer treatment for resectable PDAC would have the following characteristics: administration to the highest possible proportion of patients, ability to identify fast-progressing patients (i.e. poor candidates for surgery), a low proportion of R1 resections (through optimisation of local disease control), and an acceptable rate of toxicity profile. The neo-adjuvant approach meets these criteria and has been validated (or is being evaluated) for other main digestive tract sites (the rectum, stomach, oesophagus and colon). With respect to published data on the efficacy of FOLFOX and mFOLFIRINOX, these two combination regimens are potential candidates for neo-adjuvant use with a view to optimize oncological outcomes in resectable PDAC.
Change history
03 March 2020
Following publication of the original article [1], the authors reported an error in the “Samples size calculation and statistical considerations” section.
Abbreviations
- DFS:
-
Disease free survival
- HRQoL:
-
Health-related quality of life
- OS:
-
Overall survival
- PDAC:
-
Pancreatic duct adenocarcinoma
- PV:
-
Portal vein
- R0:
-
Disease free margin
- SMA:
-
Superior mesenteric artery
- SMV:
-
Superior mesenteric vein
References
Carpelan-Holmstrom M, Nordling S, Pukkala E, et al. Does anyone survive pancreatic ductal adenocarcinoma? A nationwide study re-evaluating the data of the Finnish Cancer registry. Gut. 2005;54:385–7.
Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49.
Guerin S, Hill C. Cancer epidemiology in France in 2010, comparison with the USA. Bulletin du cancer. 2010;97:47–54.
Simard EP, Ward EM, Siegel R, et al. Cancers with increasing incidence trends in the United States: 1999 through 2008. CA Cancer J Clin. 2012;
Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–77.
Neoptolemos JP, Stocken DD, Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350:1200–10.
Neoptolemos JP, Stocken DD, Bassi C, et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA. 2010;304:1073–81.
Murakami Y, Uemura K, Sudo T, et al. Number of metastatic lymph nodes, but not lymph node ratio, is an independent prognostic factor after resection of pancreatic carcinoma. J Am Coll Surg. 2010;211:196–204.
Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, version 2.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2017;15:1028–61.
Verbeke CS, Smith AM. Survival after pancreaticoduodenectomy is not improved by extending resections to achieve negative margins. Ann Surg. 2010;251:776–7. author reply 777–778
Esposito I, Kleeff J, Bergmann F, et al. Most pancreatic cancer resections are R1 resections. Ann Surg Oncol. 2008;15:1651–60.
Delpero JR, Bachellier P, Regenet N, et al. Pancreaticoduodenectomy for pancreatic ductal adenocarcinoma: a French multicentre prospective evaluation of resection margins in 150 evaluable specimens. HPB (Oxford). 2014;16:20-33
Ghaneh P, Kleeff J, Halloran CM, et al. The impact of positive resection margins on survival and recurrence following resection and adjuvant chemotherapy for pancreatic ductal adenocarcinoma. Ann Surg. 2017. [Epub ahead of print].
Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised. phase 3 trial Lancet. 2017;389:1011–24.
DeOliveira ML, Winter JM, Schafer M, et al. Assessment of complications after pancreatic surgery: a novel grading system applied to 633 patients undergoing pancreaticoduodenectomy. Ann Surg. 2006;244:931–7. discussion 937–939
Gouma DJ, van Geenen RC, van Gulik TM, et al. Rates of complications and death after pancreaticoduodenectomy: risk factors and the impact of hospital volume. Ann Surg. 2000;232:786–95.
Le Page S, Caputo S, Kwiatkowski F, Berard P, Gouillat C. Séquelles fonctionnelles et qualité de vie après duodénopancréatectomie céphalique. J de chirurgie. 2008;145:32–6.
Regine WF, Winter KA, Abrams RA, et al. Fluorouracil vs gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: a randomized controlled trial. JAMA. 2008;299:1019–26.
Oettle H, Neuhaus P, Hochhaus A, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA. 2013;310:1473–81.
Gillen S, Schuster T, Meyer Zum Buschenfelde C, et al. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010;7:e1000267.
Tzeng CW, Tran Cao HS, Lee JE, et al. Treatment sequencing for resectable pancreatic cancer: influence of early metastases and surgical complications on multimodality therapy completion and survival. J Gastrointest Surg. 2014;18:16–24. discussion 24–15
Merkow RP, Bilimoria KY, Tomlinson JS, et al. Postoperative complications reduce adjuvant chemotherapy use in Resectable pancreatic Cancer. Ann Surg. 2014;260:372-7.
Valle JW, Palmer D, Jackson R, et al. Optimal duration and timing of adjuvant chemotherapy after definitive surgery for ductal adenocarcinoma of the pancreas: ongoing lessons from the ESPAC-3 study. J Clin Oncol. 2014;32:504–12.
Ueno H, Kosuge T, Matsuyama Y, et al. A randomised phase III trial comparing gemcitabine with surgery-only in patients with resected pancreatic cancer: Japanese study Group of Adjuvant Therapy for pancreatic Cancer. Br J Cancer. 2009;101:908–15.
Hoffman JP, Lipsitz S, Pisansky T, et al. Phase II trial of preoperative radiation therapy and chemotherapy for patients with localized, resectable adenocarcinoma of the pancreas: an Eastern Cooperative Oncology Group Study. J Clin Oncol. 1998;16:317–23.
Motoi F, Ishida K, Fujishima F, et al. Neoadjuvant chemotherapy with gemcitabine and S-1 for Resectable and borderline pancreatic ductal adenocarcinoma: results from a prospective multi-institutional phase 2 trial. Ann Surg Oncol. 2013;20(12):3794–801.
Takahashi H, Ohigashi H, Gotoh K, et al. Preoperative gemcitabine-based Chemoradiation therapy for Resectable and borderline Resectable pancreatic Cancer. Ann Surg. 2013;258:1040-50.
Palmer DH, Stocken DD, Hewitt H, et al. A randomized phase 2 trial of neoadjuvant chemotherapy in resectable pancreatic cancer: gemcitabine alone versus gemcitabine combined with cisplatin. Ann Surg Oncol. 2007;14:2088–96.
Varadhachary GR, Wolff RA, Crane CH, et al. Preoperative gemcitabine and cisplatin followed by gemcitabine-based chemoradiation for resectable adenocarcinoma of the pancreatic head. J Clin Oncol. 2008;26:3487–95.
Evans DB, Varadhachary GR, Crane CH, et al. Preoperative gemcitabine-based chemoradiation for patients with resectable adenocarcinoma of the pancreatic head. J Clin Oncol. 2008;26:3496–502.
White RR, Hurwitz HI, Morse MA, et al. Neoadjuvant chemoradiation for localized adenocarcinoma of the pancreas. Ann Surg Oncol. 2001;8:758–65.
Reni M, Balzano G, Aprile G, et al. Adjuvant PEFG (cisplatin, epirubicin, 5-fluorouracil, gemcitabine) or gemcitabine followed by chemoradiation in pancreatic cancer: a randomized phase II trial. Ann Surg Oncol. 2012;19:2256–63.
Picozzi VJ, Abrams RA, Decker PA, et al. Multicenter phase II trial of adjuvant therapy for resected pancreatic cancer using cisplatin, 5-fluorouracil, and interferon-alfa-2b-based chemoradiation: ACOSOG trial Z05031. Ann Oncol. 2011;22:348–54.
Takahashi H, Ohigashi H, Gotoh K, et al. Preoperative gemcitabine-based chemoradiation therapy for resectable and borderline resectable pancreatic cancer. Ann Surg. 2013;258:1040–50.
Mokdad AA, Minter RM, Zhu H, et al. Neoadjuvant therapy followed by resection versus upfront resection for Resectable pancreatic Cancer: a propensity score matched analysis. J Clin Oncol. 2017;35:515–22.
Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol. 2016;17:801–10.
Rombouts SJ, Walma MS, Vogel JA, et al. Systematic review of resection rates and clinical outcomes after FOLFIRINOX-based treatment in patients with locally advanced pancreatic Cancer. Ann Surg Oncol. 2016;23:4352–60.
Pietrasz D, Marthey L, Wagner M, et al. Pathologic major response after FOLFIRINOX is prognostic for patients secondary resected for borderline or locally advanced pancreatic adenocarcinoma: an AGEO-FRENCH, prospective, multicentric cohort. Ann Surg Oncol. 2015;22(Suppl 3):S1196–205.
Ferrone CR, Marchegiani G, Hong TS, et al. Radiological and surgical implications of neoadjuvant treatment with FOLFIRINOX for locally advanced and borderline resectable pancreatic cancer. Ann Surg. 2015;261:12–7.
Ghosn M, Saroufim A, Kattan J, et al. Sequential FOLFOX-6 and gemcitabine for locally advanced and/or metastatic pancreatic cancer. Med Oncol. 2012;29:2831–7.
Oettle H, Riess H, Stieler JM, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. J Clin Oncol. 2014;32:2423–9.
Bonnetain F, Bonsing B, Conroy T, et al. Guidelines for time-to-event end-point definitions in trials for pancreatic cancer. Results of the DATECAN initiative (definition for the assessment of time-to-event end-points in CANcer trials). Eur J Cancer. 2014;50:2983–93.
Bellera CA, Pulido M, Gourgou S, et al. Protocol of the definition for the assessment of time-to-event endpoints in CANcer trials (DATECAN) project: formal consensus method for the development of guidelines for standardised time-to-event endpoints' definitions in cancer clinical trials. Eur J Cancer. 2013;49:769–81.
Fitzsimmons D, Johnson CD, George S, et al. Development of a disease specific quality of life (QoL) questionnaire module to supplement the EORTC core cancer QoL questionnaire, the QLQ-C30 in patients with pancreatic cancer. EORTC Study Group on Quality of Life. Eur J Cancer. 1999;35:939–41.
Dindo D, Demartines N, Clavien PA. Classification of surgical complications: a new proposal with evaluation in a cohort of 6336 patients and results of a survey. Ann Surg. 2004;240:205–13.
Bassi C, Dervenis C, Butturini G, et al. Postoperative pancreatic fistula: an international study group (ISGPF) definition. Surgery. 2005;138:8–13.
Kim HW, Lee JC, Paik KH, et al. Adjunctive role of preoperative liver magnetic resonance imaging for potentially resectable pancreatic cancer. Surgery. 2017;161:1579–87.
Jeon SK, Lee JM, Joo I, et al. Magnetic resonance with diffusion-weighted imaging improves assessment of focal liver lesions in patients with potentially resectable pancreatic cancer on CT. Eur Radiol. 2018;28(8):3484–93.
van der Gaag NA, Rauws EA, van Eijck CH, et al. Preoperative biliary drainage for cancer of the head of the pancreas. N Engl J Med. 2010;362:129–37.
Tol JA, van Hooft JE, Timmer R, et al. Metal or plastic stents for preoperative biliary drainage in resectable pancreatic cancer. Gut. 2016;65:1981–7.
Slim K, Blay JY, Brouquet A, et al. Digestive oncology: surgical practices. J Chir. 2009;146(Suppl 2):S11–80.
Mitchem JB, Hamilton N, Gao F, et al. Long-term results of resection of adenocarcinoma of the body and tail of the pancreas using radical antegrade modular pancreatosplenectomy procedure. J Am Coll Surg. 2012;214:46–52.
Verbeke CS, Menon KV. Redefining resection margin status in pancreatic cancer. HPB (Oxford). 2009;11:282–9.
Bryant J, Day R. Incorporating toxicity considerations into the design of two-stage phase II clinical trials. Biometrics. 1995;51:1372–83.
Klinkenbijl JH, Jeekel J, Sahmoud T, et al. Adjuvant radiotherapy and 5-fluorouracil after curative resection of cancer of the pancreas and periampullary region: phase III trial of the EORTC gastrointestinal tract cancer cooperative group Ann Surg 1999: 230; 776–782; discussion 782–774.
Spitz FR, Abbruzzese JL, Lee JE, et al. Preoperative and postoperative chemoradiation strategies in patients treated with pancreaticoduodenectomy for adenocarcinoma of the pancreas. J Clin Oncol. 1997;15:928–37.
Corsini MM, Miller RC, Haddock MG, et al. Adjuvant radiotherapy and chemotherapy for pancreatic carcinoma: the Mayo Clinic experience (1975-2005). J Clin Oncol. 2008;26:3511–6.
Herman JM, Swartz MJ, Hsu CC, et al. Analysis of fluorouracil-based adjuvant chemotherapy and radiation after pancreaticoduodenectomy for ductal adenocarcinoma of the pancreas: results of a large, prospectively collected database at the Johns Hopkins Hospital. J Clin Oncol. 2008;26:3503–10.
Schorn S, Demir IE, Reyes CM, et al. The impact of neoadjuvant therapy on the histopathological features of pancreatic ductal adenocarcinoma - a systematic review and meta-analysis. Cancer Treat Rev. 2017;55:96–106.
Kanda M, Fujii T, Sahin TT, et al. Invasion of the splenic artery is a crucial prognostic factor in carcinoma of the body and tail of the pancreas. Ann Surg. 2010;251:483–7.
Schnelldorfer T, Ware AL, Sarr MG, et al. Long-term survival after pancreatoduodenectomy for pancreatic adenocarcinoma: is cure possible? Ann Surg. 2008;247:456–62.
Rajagopalan MS, Heron DE, Wegner RE, et al. Pathologic response with neoadjuvant chemotherapy and stereotactic body radiotherapy for borderline resectable and locally-advanced pancreatic cancer. Radiat Oncol. 2013;8:254.
Katz MH, Shi Q, Ahmad SA, et al. Preoperative modified FOLFIRINOX treatment followed by Capecitabine-based Chemoradiation for borderline Resectable pancreatic Cancer: alliance for clinical trials in oncology trial A021101. JAMA Surg. 2016;151:e161137.
Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–13.
Maisey N, Chau I, Cunningham D, et al. Multicenter randomized phase III trial comparing protracted venous infusion (PVI) fluorouracil (5-FU) with PVI 5-FU plus mitomycin in inoperable pancreatic cancer. J Clin Oncol. 2002;20:3130–6.
Colucci G, Giuliani F, Gebbia V, et al. Gemcitabine alone or with cisplatin for the treatment of patients with locally advanced and/or metastatic pancreatic carcinoma: a prospective, randomized phase III study of the Gruppo Oncologia dell'Italia Meridionale. Cancer. 2002;94:902–10.
Colucci G, Labianca R, Di Costanzo F, et al. Randomized phase III trial of gemcitabine plus cisplatin compared with single-agent gemcitabine as first-line treatment of patients with advanced pancreatic cancer: the GIP-1 study. J Clin Oncol. 2010;28:1645–51.
Heinemann V, Wilke H, Mergenthaler HG, et al. Gemcitabine and cisplatin in the treatment of advanced or metastatic pancreatic cancer. Ann Oncol. 2000;11:1399–403.
Louvet C, Labianca R, Hammel P, et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J Clin Oncol. 2005;23:3509–16.
Poplin E, Feng Y, Berlin J, et al. Phase III, randomized study of gemcitabine and oxaliplatin versus gemcitabine (fixed-dose rate infusion) compared with gemcitabine (30-minute infusion) in patients with pancreatic carcinoma E6201: a trial of the eastern cooperative oncology group. J Clin Oncol. 2009;27:3778–85.
Wagner AD, Buechner-Steudel P, Wein A, et al. Gemcitabine, oxaliplatin and weekly high-dose 5-FU as 24-h infusion in chemonaive patients with advanced or metastatic pancreatic adenocarcinoma: a multicenter phase II trial of the Arbeitsgemeinschaft Internistische Onkologie (AIO). Ann Oncol. 2007;18:82–7.
Novarino A, Chiappino I, Bertelli GF, et al. Phase II study of cisplatin, gemcitabine and 5-fluorouracil in advanced pancreatic cancer. Ann Oncol. 2004;15:474–7.
Hess V, Pratsch S, Potthast S, et al. Combining gemcitabine, oxaliplatin and capecitabine (GEMOXEL) for patients with advanced pancreatic carcinoma (APC): a phase I/II trial. Ann Oncol. 2010;21:2390–5.
Correale P, Montagnani F, Miano S, et al. Biweekly triple combination chemotherapy with gemcitabine, oxaliplatin, levofolinic acid and 5-fluorouracil (GOLF) is a safe and active treatment for patients with inoperable pancreatic cancer. J Chemother. 2008;20:119–25.
Conroy T, Paillot B, Francois E, et al. Irinotecan plus oxaliplatin and leucovorin-modulated fluorouracil in advanced pancreatic cancer--a Groupe Tumeurs digestives of the federation Nationale des Centres de Lutte Contre le Cancer study. J Clin Oncol. 2005;23:1228–36.
Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. New England J medicine. 2011;364:1817–25.
Gunturu KS, Yao X, Cong X, et al. FOLFIRINOX for locally advanced and metastatic pancreatic cancer: single institution retrospective review of efficacy and toxicity. Med Oncol. 2013;30(361)
Marthey L, Sa-Cunha A, Blanc JF, et al. FOLFIRINOX for locally advanced pancreatic adenocarcinoma: results of an AGEO multicenter prospective observational cohort. Ann Surg Oncol. 2015;22:295-301.
Goel A, Grossbard ML, Malamud S, et al. Pooled efficacy analysis from a phase I-II study of biweekly irinotecan in combination with gemcitabine, 5-fluorouracil, leucovorin and cisplatin in patients with metastatic pancreatic cancer. Anti-Cancer Drugs. 2007;18:263–71.
Reni M, Sartori N, Mambrini A, et al. An Italian study on treatment trends and outcomes of patients with stage III pancreatic adenocarcinoma in the gemcitabine era: is it time to change? Anti-Cancer Drugs. 2010;21:459–64.
Farrell JJ, Elsaleh H, Garcia M, et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology. 2009;136:187–95.
Marechal R, Bachet JB, Mackey JR, et al. Levels of gemcitabine transport and metabolism proteins predict survival times of patients treated with gemcitabine for pancreatic adenocarcinoma. Gastroenterology. 2012;143:664–74. e661–666
Bird NT, Elmasry M, Jones R, et al. Immunohistochemical hENT1 expression as a prognostic biomarker in patients with resected pancreatic ductal adenocarcinoma undergoing adjuvant gemcitabine-based chemotherapy. Br J Surg. 2017;104:328–36.
Yamada R, Mizuno S, Uchida K, et al. Human Equilibrative nucleoside transporter 1 expression in endoscopic ultrasonography-guided fine-needle aspiration biopsy samples is a strong predictor of clinical response and survival in the patients with pancreatic ductal adenocarcinoma undergoing gemcitabine-based Chemoradiotherapy. Pancreas. 2016;45:761–71.
Lutz MP, Zalcberg JR, Ducreux M, et al. 3rd St. Gallen EORTC gastrointestinal Cancer conference: consensus recommendations on controversial issues in the primary treatment of pancreatic cancer. Eur J Cancer. 2017;79:41–9.
Hosein PJ, Macintyre J, Kawamura C, et al. A retrospective study of neoadjuvant FOLFIRINOX in unresectable or borderline-resectable locally advanced pancreatic adenocarcinoma. BMC Cancer. 2012;12(199)
Faris JE, Blaszkowsky LS, McDermott S, et al. FOLFIRINOX in locally advanced pancreatic cancer: the Massachusetts General Hospital Cancer center experience. Oncologist. 2013;18:543–8.
Ducreux M, Mitry E, Ould-Kaci M, et al. Randomized phase II study evaluating oxaliplatin alone, oxaliplatin combined with infusional 5-FU, and infusional 5-FU alone in advanced pancreatic carcinoma patients. Ann Oncol. 2004;15:467–73.
Ghosn M, Farhat F, Kattan J, et al. FOLFOX-6 combination as the first-line treatment of locally advanced and/or metastatic pancreatic cancer. Am J Clin Oncol. 2007;30:15–20.
Gebbia V, Maiello E, Giuliani F, et al. Second-line chemotherapy in advanced pancreatic carcinoma: a multicenter survey of the Gruppo Oncologico Italia Meridionale on the activity and safety of the FOLFOX4 regimen in clinical practice. Ann Oncol. 2007;18(Suppl 6):vi124–7.
Novarino A, Satolli MA, Chiappino I, et al. Oxaliplatin, 5-fluorouracil, and leucovorin as second-line treatment for advanced pancreatic cancer. Am J Clin Oncol. 2009;32:44–8.
Pelzer U, Schwaner I, Stieler J, et al. Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III-study from the German CONKO-study group. Eur J Cancer. 2011;47:1676–81.
Yoo C, Hwang JY, Kim JE, et al. A randomised phase II study of modified FOLFIRI.3 vs modified FOLFOX as second-line therapy in patients with gemcitabine-refractory advanced pancreatic cancer. Br J Cancer. 2009;101:1658–63.
Zaanan A, Trouilloud I, Markoutsaki T, et al. FOLFOX as second-line chemotherapy in patients with pretreated metastatic pancreatic cancer from the FIRGEM study. BMC Cancer. 2014;14(441)
Acknowledgments
We thank all the cooperative groups (FFCD – UNICANCER – FRENCH – GERCOR) for their contribution and participation to the present trial. We warmly thank Franck Bonnetain for his participation in the development of this project. We also thank all the participating centers.
Thirty four French centres will participate in the study: Amiens University Hospital, Angers University Hospital, University Hospital of Clermont-Ferrand, European Georges Pompidou University Hospital in Paris, Claude Huriez University Hospital in Lille, University hospital of Bordeaux, University Hospital in Toulouse, Paoli Calmettes Cancer Institute in Marseille, Henri Mondor University Hospital in Créteil, North University Hospital in Marseille, Pitié Salpêtrière University Hospital in Paris, Croix-Rousse University Hospital in Lyon, Beaujon University hospital in Clichy, the University Hospital in Strasbourg, Pontchaillou University Hospital in Rennes, Robert Debré University Hospital in Reims, Cochin University Hospital in Paris, University Hospital of Nantes, La Timone University Hospital in Marseille, Saint Antoine University Hospital in Paris, University Hospital of Dijon, University Hospital of Rouen, Eugene Marquis Cancer Institut of Rennes, Paul Brousse University Hospital in Villejuif, Montpellier University Hospital, Gustave Roussy in Villejuif, Institut Mutualiste Montsouris in Paris, Tours University Hospital, Caen University hospital, Edouard Herriot University Hospital in Lyon, Kremlin Bicetre University Hospital, Nancy University Hospital, Poitiers University Hospital, Tours University Hospital.
Funding
This project was funded by the Programme Hospitalier de Recherche Clinique from the French National Cancer Institute (INCa) in 2014. The funding will aim to enable data collection and analysis, interpretation of data and writing manuscript. The funding body has no role in research design, data acquisition or analysis, or in writing manuscript.
Author information
Authors and Affiliations
Contributions
All authors are rightly credited with authorship. Conception and design: LS, ASC. Methodology: DV. Drafting of the manuscript: LS, ASC. Critical revision of the manuscript: ASC, DV, JJT, PM, FM, JBB. Final approval: LS, DV, ASC, JJT, PM, FM, JBB.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Institutional Review Board, the Nord Ouest IV ethics committee on the 26th of May 2016 and the Agence Nationale de sécurité des médicaments et des produits de santé (ANSM) on the 19th of July 2016 under the number 2015–001851-65. The institutional promoter is the University Hospital of Rouen, France. The trial has been registered on the ClinicalTrial.gov website under the identification number NCT02959879, on November 9, 2016. The study complies with the Declaration of Helsinki rules and the principles of Good Clinical Practice guidelines. Informed consent will be obtained from each patient in written form prior to randomization and an information leaflet will be given to each patient prior to inclusion in the study. Patient safety and all potential threats to patient safety will be monitored by an independent monitoring board which will be able, taking into account undesirable events and the level of post-operative mortality, recommend the discontinuation of one of the treatments evaluated.
Consent for publication
Not applicable
Competing interests
None of the authors have any competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Schwarz, L., Vernerey, D., Bachet, JB. et al. Resectable pancreatic adenocarcinoma neo-adjuvant FOLF(IRIN)OX-based chemotherapy - a multicenter, non-comparative, randomized, phase II trial (PANACHE01-PRODIGE48 study). BMC Cancer 18, 762 (2018). https://doi.org/10.1186/s12885-018-4663-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-018-4663-4