
Abstract
Background
Head and neck squamous cell carcinoma (HNSCC) is a heterogeneous group of tumours with a typical 5 year survival rate of <40 %. DNA methylation in tumour-suppressor genes often occurs at an early stage of tumorigenesis, hence DNA methylation can be used as an early tumour biomarker. Saliva is an ideal diagnostic medium to detect early HNSCC tumour activities due to its proximity to tumour site, non-invasiveness and ease of sampling. We test the hypothesis that the surveillance of DNA methylation in five tumour-suppressor genes (RASSF1α, p16INK4a, TIMP3, PCQAP/MED15) will allow us to diagnose HNSCC patients from a normal healthy control group as well as to discriminate between Human Papillomavirus (HPV)-positive and HPV-negative patients.
Methods
Methylation-specific PCR (MSP) was used to determine the methylation levels of RASSF1α, p16INK4a, TIMP3 and PCQAP/MED15 in DNA isolated from saliva. Statistical analysis was carried out using non-parametric Mann-Whitney’s U-test for individually methylated genes. A logistic regression analysis was carried out to determine the assay sensitivity when combing the five genes. Further, a five-fold cross-validation with a bootstrap procedure was carried out to determine how well the panel will perform in a real clinical scenario.
Results
Salivary DNA methylation levels were not affected by age. Salivary DNA methylation levels for RASSF1α, p16INK4a, TIMP3 and PCQAP/MED15 were higher in HPV-negative HNSCC patients (n = 88) compared with a normal healthy control group (n = 122) (sensitivity of 71 % and specificity of 80 %). Conversely, DNA methylation levels for these genes were lower in HPV-positive HNSCC patients (n = 45) compared with a normal healthy control group (sensitivity of 80 % and specificity of 74 %), consistent with the proposed aetiology of HPV-positive HNSCCs.
Conclusions
Salivary DNA tumour-suppressor methylation gene panel has the potential to detect early-stage tumours in HPV-negative HNSCC patients. HPV infection was found to deregulate the methylation levels in HPV-positive HNSCC patients. Large-scale double-blinded clinical trials are crucial before this panel can potentially be integrated into a clinical setting.
Similar content being viewed by others


Background
Head and neck squamous cell carcinomas (HNSCCs) encompasses tumours within the oral cavity, pharynx, larynx, paranasal sinuses, nasal cavity and salivary glands, and are some of the most aggressive cancer types [1, 2]. Main risk factors for HNSCC include smoking, alcohol consumption, betel quid chewing, and Human Papillomavirus (HPV, mainly HPV-16 and HPV-18) infections [2–4]. HPV-positive and HPV-negative cancers are biologically and clinically different and as such require different treatment and clinical management [5].
HNSCC is the sixth most common cancer worldwide with ~780,000 new cases diagnosed each year [6]. The incidence of HNSCC in developed countries has decreased over the past 20 years, which is largely attributed to a reduction in smoking and alcohol consumption [4]. However, the incidence of HPV-positive HNSCC is on the rise and accounts for 30 to 50 % of all HNSCCs [4, 6]. HPV-positive HNSCC patients have cancers that are almost exclusively located in the oropharynx [7–10]. HPV-positive HNSCC patients are often young with a higher socioeconomic status and typically non-smokers [7–10]. The five-year survival rates for HPV-negative HNSCC when diagnosed early is 80 % compared with only 15 % for the advanced stage cancers [11–13].
Currently, diagnosis relies on the direct examination of the head and neck regions and is usually made after clinical presentation of symptoms and involves biopsy to confirm diagnosis. The HPV status of a patient is determined by p16INK4a immunohistochemistry (IHC) staining on tumour tissue samples and histological classification using the tumour-node-metastasis (TNM) [14–16]. Direct examination is highly-subjective and becomes problematic when tumours are too small to be visualised, or are hidden in obscure areas such as the tonsillar crypts or within the pits and crevices in the lingual tonsils of the tongue base. This would then likely require techniques such as nasendoscopy or examination under anaesthesia to locate the tumour and both require biopsy for confirmation. These issues may commonly result in misdiagnosis [17]. The direct contact between saliva and oral cavity lesions make the measurement of the tumour markers in saliva an attractive alternative to serum and tumour tissue biopsy testings [6, 18–21]. Saliva is now championed as the diagnostic fluid of the future over blood and urine as saliva testing is easy, inexpensive, safe, and non-invasive [19, 22–25].
Gene-specific DNA methylation, especially in tumour-suppressor genes, is recognized as a contributor to the regulation of gene expression and phenotypic heterogeneity in HNSCC [26, 27]. The DNA promoter methylation analysis in saliva samples collected from HNSCC patients have previously been shown to demonstrate clinical utility [6, 25, 28]. The most commonly used method for the detection of DNA methylation analysis in tissue and body fluids is the methylation-specific PCRs (MSPs) [29]. MSP analysis is highly sensitive and does not require expensive laboratory equipment and is therefore economical compared to other quantitative DNA methylation analysis such as pyrosequencing and real-time quantitate MSP [30, 31]. In addition, MSP is able to provide a time-efficient and direct DNA methylation status analysis, making it convenient for large-scale sample screening [30, 31]. The ability to relatively quantify DNA methylation signatures allows the delineation of clinically meaningful threshold values to discriminate a patient cohort from a control cohort.
We hypothesise that by analysing DNA methylation of tumour-suppressor genes in saliva; we can detect early tumour activities as well as to differentially diagnose HNSCC patients. Our study objectives are two-fold: (i) firstly, to investigate the early diagnostic potential of the salivary DNA methylation panel (RASSF1α, p16INK4a, TIMP3, PCQAP 5′ and PCQAP 3′) (ii) secondly, to determine whether this panel is able to discriminate between HPV-negative and HPV-positive HNSCC patients. We selected this panel as we have previously published individual DNA methylation levels in saliva collected from HNSCC patient and controls except for TIMP3. From our previously published work, we were able to discriminate normal healthy controls from HNSCC patients using these individual DNA methylation levels [6, 25]. In this study, we have combined the DNA methylation levels for all of the five tumour-suppressor genes as a panel to increase the sensitivity and specificity when discriminating normal healthy controls from HNSCC patients. Our salivary DNA methylation panel is able to detect HPV-negative HNSCC patients from a normal healthy control group with a sensitivity of 71 % and specificity of 80 %. In contrast, the DNA methylation levels were lower in saliva collected from HPV-positive HNSCC patients compared with normal healthy controls (sensitivity of 80 % and specificity of 74 %). It is important to conduct a multi-centre clinical trial before this panel can be implemented in a clinical setting.
Methods
Study design
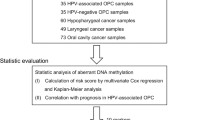
This study is approved by the University of Queensland (HREC no.: 2014000679) and Queensland University of Technology (HREC no.: 1400000617) Medical Ethical Institutional Boards and the Princess Alexandra Hospital’s (PAH) Ethics Review Board (HREC no.: HREC/12/QPAH/381). We have recruited normal healthy controls, both smokers and non-smokers (n = 122) and HNSCC patients (n = 133). HNSCC patient cohort consisted of HPV-negative and HPV-positive patients. The Table 1 presents the demographic and clinical characteristics of our study cohort.
Determination of HPV-16 status in tumour samples
We obtained a pathology report for each patient which contained tumour staging information, histopathological grading and HPV-16 status. HPV-16 status was determined by staining for p16INK4a in tumour tissue section using IHC (CINtec® Histology Kit, Roche MTM Laboratories, Heidelberg, Germany) according to the manufacturer’s protocol [32]. p16 INK4a IHC was evaluated by trained pathologists [32]. The determination of HPV-16 status at the PAH is restricted to patients with cancers in the oropharynx because of the low prevalence of HPV-16 among non-oropharynx sites [9]. Therefore, p16INK4a IHC is not requested by the treating clinician when tumours are outside of the oropharyngeal area.
Saliva sample collection and processing
In the clinic, volunteers were asked to refrain from eating and drinking for an hour prior to donating saliva samples. The volunteers were asked to sit in a comfortable position and were asked to rinse their mouths with water to remove food debris. They were then asked to pool saliva in the mouth and expectorate directly into a 50 mL Falcon tube. Saliva samples were transported from the hospital to the laboratory on dry ice. Samples were centrifuged at 1500 × g for 10 min at 4 °C, separating cellular pellet from cell-free salivary supernatant. Cellular pellet was used to isolate DNA, which was subsequently subjected to bisulfite conversion.
DNA extraction and bisulfite conversion from saliva samples
The Epitect® Plus DNA Bisulfite Kit (Cat. No. 59124, Qiagen, Duesseldorf, Germany) was used to extract and bisulfite-convert DNA from salivary cellular pellet according to the manufacturer’s protocol. An additional 10 min of incubation time was adapted due to a change in elution volume of 17 μL instead of 15 μL. Purity and quantity of the converted DNA samples were measured with a Nano Drop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
Methylation-specific PCR assays
The MSP primer pairs (RASSF1a, p16INK4a, TIMP3) used in this study has been extensively validated in other studies, except for MED15/PCQAP [6, 25]. MED15/PCQAP novel CpG sites were identified by our group and we have previously confirmed the specificity of amplicons using the MSP primer pairs and we have also verified the PCR amplicon sequence (Additional file 1: Figure S1) [25]. The primer specificities for RASSF1a and p16INK4a were confirmed by Divine et al., 2006 using the denaturing high performance liquid chromatography (DHPLC) [6, 33]. Similarly, TIMP3 MSP primer pairs was initially used in a MethyLight assay by Eads et al. in 2001 and later modified by Righini et al. to be compatible with a MSP assay [34, 35].
To determine the specificity of the MSP primers, all MSP primers (both methylation and unmethylation) were tested using bisulfite unconverted DNA samples and was found not to amplify. This proves the specificity of the primer pairs used in this study. Unmethylation PCRs were used as a normaliser for methylation PCRs. Samples without unmethylation bands were either discarded from the analysis or repeated. Bisulfite-treated methylated HeLa cell line DNA (Cat. No.4007s, New England Biolabs, Ipswich, Massachusetts, USA) was used as a positive control while DNase/RNase-free distilled water (blank) was used as a negative control for the MSP assays.
The quantitative nature and efficiency of conventional MSP was established by using bisulfite-treated methylated HeLa cells at varying amounts. In brief, HeLa cells were spiked in oral adenosquamous carcinoma cell line, (CAL27) in a six-point serial dilution format to generate a standard curve using the ratio of methylation to unmethylation PCR reactions (Fig. 1) [36]. Our results clearly demonstrate that the conventional MSP is a reliable way to relatively quantify methylation levels (MSP efficiencies of >0.8) (Fig. 1).

A six-point standard curve spiking of positive cell line, HeLa in oral adenosquamous cell carcinoma, CAL27 of a RASSF1α, b p16INK4a, c TIMP3, d PCQAP 5′ and e PCQAP 3′
RASSF1α and p16INK4a were amplified using nested MSP. Nested MSP primer sets for both stage-1 (nested, methylation-insensitive stage) and 2 (methylation-sensitive stage) are presented in Table 2 [6]. Briefly, stage-1 PCR amplification for RASSF1α and p16INK4a was carried out using 1 μM of the appropriate nested primer sets, 6.25 μL of EmeraldAmp® MAX HS PCR Master Mix (TaKaRa Bio Inc., Otsu, Shiga, Japan) and 1.25 ng and 20 ng of DNA template respectively. The total reaction volume of 12.5 μL was subjected to PCR amplification using the following conditions: initial denaturing stage at 94 °C for two minutes, followed by 30 cycles of 15 s at 94 °C, 15 s at 60 °C and 15 s at 72 °C. In stage-2, two corresponding sets of methylated and unmethylated primers for each gene were used. The amplification cycling conditions included: initial denaturing stage at 94 °C for 2 min, followed by 5 cycles of 15 s at 94 °C, 15 s at 62 °C and 15 s at 72 °C with three repeats of decreasing annealing temperature (64, 62 and 60 °C in that order) before extension stage at 72 °C for 5 min. Stage-2 PCRs used 1 μL of stage-1 product as DNA template.
For TIMP3, unique methylated and unmethylated primer sets for each gene was used to target their corresponding CpG-methylation sites (Table 2) [34]. The PCR reaction consisted of 5 μL of EmeraldAmp® MAX HS PCR Master Mix and 0.8 μM of their respective primer sets, in 10 μL final reaction volume. Total DNA template ratio of 20:1 was used for the methylated reaction and unmethylated reaction respectively. The PCR amplification consisted of initial denaturing stage at 95 °C for 5 min, followed by 40 cycles of 15 s at 94 °C, 15 s at 54 °C and 15 s at 72 °C before summing up with elongation stage at 72 °C for 4 min.
Similar to TIMP3, PCQAP (Table 2) also required two separate setup conditions for the methylated and unmethylated reactions under the same cycling condition. Both methylated and unmethylatd reactions consisted of 6.25 μL of EmeraldAmp® MAX HS PCR Master Mix and 1 μM of their respective primer sets. In terms of DNA template concentrations, ratio of 25:1 was used for the methylated reactions and unmethylated reactions respectively. The PCR amplification consisted of initial denaturing stage at 95 °C for 3 min, followed by 35 cycles of 30 s at 94 °C, 30 s at 62.5 °C and 1 min at 72 °C before summing up with elongation stage at 72 °C for 5 min. PCQAP MSP reactions required an addition of 5 % DMSO and 0.1 μg/mL BSA to minimise the presence of unspecific bands caused by secondary DNA structures [25].
Gel electrophoresis and densitometry analysis
MSP analysis was carried out by running 5 μL PCR amplicon products on 2 % agarose gel. The gels were scanned on Fusion SL (Vilber Lourmat, Marne la Vallee, France) and visualized using ImageJ software (National Institutes of Health, Bethesda, Maryland, USA). In order to quantify the ratio between methylated and unmethylated bands, samples with saturated bands were re-run with a lower concentration ratio of DNA template for both methylated and unmethylated PCRs.
The methylated and unmethylated band intensities were quantified using ImageJ software and the ratio between methylated to unmethylated was calculated for each sample using Microsoft Excel (Microsoft Corporation, Redmond, Washington, USA). A standard rectangular-frame was estimated according to the size of the smallest band in a given gel. Consequently, the same rectangular-frame was used to measure the intensity of each band within the same gel to provide consistency. The measurement was set at integrated density to calculate the intensity value of the band based on the amount of amplicon present. All quantifications were carried out by two independent researchers to minimise observational errors.
Statistical analysis
The statistical analysis was carried out by using GraphPad Prism (GraphPad Software, Inc, San Diego, California, USA) and R (R.D.C. Team, Vienna, Austria). The methylation levels were not normally distributed and therefore a non-parametric test (Mann-Whitney U test) was used when comparing the data generated using normal healthy controls with HPV-negative and HPV-positive HNSCC patients respectively. In addition, Spearman’s rank correlation test was used to determine the correlation between patients’ age and methylation level given that age is a continuous variable.
The overarching aim of this study is to evaluate the diagnostic potential of the combined five tumour-suppressor genes in a panel and as such, the sensitivity and specificity were estimated. For this purpose, the ‘Epi’ package was used in R [37]. The patient status is used as the outcome variable and the methylation level for each gene is used as the explanatory variables in a multivariable logistic regression (Carstensen’s multivariate ROC curve). Predicted scores are then produced for each patient using the estimated regression model and different cut-off values of this predicted score are used for classifying samples into patients or controls. A known issue in this case is that the predicted classification of the samples is optimal since the same sample that has been used for creating the predicting model and for validating it. One good solution to address this type of issue is known as cross validation, the idea of which that proportion of the sample is used for creating the predictive model, and the remaining samples are used for validating the model [38–40]. In this case, a version of five-fold cross-validation was used. This is crucial to see how well the panel translates into clinical diagnosis. To make best use of our data, a bootstrap procedure was also incorporated [38–40]. With this statistical method, random samples are created by sampling with replacement from the original sample. The advantage of this technique is that the confidence intervals produced are more realistic compared to the parametric, asymptotic ones. Furthermore, this was done in a stratified manner; classifying on patient status in order to retained the original samples’ characteristics. Therefore, this procedure could be called a stratified bootstrap ROC with cross-validation. A custom written code was used to implement this in R using the above function from R. The program was ultimately run for 5000 times to include all possible combinations of predictive model available. The maximum sum of sensitivity and specificity was used to determine the best cut-off point for the panel.
TCGA data portal
To investigate the tumour methylation status of the five genes, we downloaded The Cancer Genome Atlas (TCGA) data for HNSCC tumours and normal tissues (https://tcga-data.nci.nih.gov/tcga/). HPV status annotation was available for 268 tumours profiled by Tang et al., (DOI:10.1038/ncomms3513; Additional file 2: Table S1) [41]. Tumours were grouped as HPV-positive HNSCC (n = 44), HPV-negative HNSCC (n = 223), or normal tissue samples (n = 50). Our approach was to select probes that overlapped within the CpG sites flanking our primer pairs used in our MSP assays (Additional file 3: Figure S2). Probes for RASSF1α, TIMP3 and PCQAP were extracted and the DNA methylation values for these three groups were plotted in R. However, there were no probes that overlapped or positioned adjacent to the CpG methylation sites interrogated by our p16INK4a MSP assays. As such, we were unable to present TCGA data for p16INK4a.
Results
Population characteristics
The mean age for normal healthy controls was 50 years (SD: 8.4 years), and consisted of 44.3 % men and 55.7 % women (Table 1). The mean age for HNSCC patients was 64 years (SD: 12.2 years), and consisted of 82.0 % men and 18.0 % women (Table 1). Cancer sites were mostly of oropharyngeal and oral cavity (53.4 and 37.6 % respectively) while laryngeal and neck cancers made up about 7.5 % of cases with only 1.5 % of cases were hypopharyngeal. In addition, 27.1 % of cases were stage I and II, whilst 54.9 % of cases were stages III and IV (Table 1). Within the HNSCC patient cohort, 4.5 % of patients were classified as current smokers, or having quit within the past 12 months, while 47.4 % were former smokers (quit more than one year ago) and 19.5 % have never smoked (Table 1). Although we do not have all the patient information regarding alcohol consumption, most of the recruited patients were alcohol users (71 %) (Table 1).
HPV-positive HNSCC patients (n = 45) were on average younger than HPV-negative HNSCC patients (n = 88) (mean age: 60 years, SD: 10.4 years, for HPV-positive HNSCC patients and mean age: 66 years, SD: 12.6 years for HPV-negative HNSCC patients, p < 0.0001) (Table 1). There were significantly more men than women patients by HPV status (93.3 % men in HPV-positive HNSCC cohort; 76.1 % men in HPV-negative HNSCC cohort, p < 0.0001) (Table 1). The majority of HPV-negative HNSCC patients had cancers within the oral cavity (76.1 %) whereas the majority of HPV-positive HNSCC patients had cancers in the oropharynx (86.7 %) (Table 1). Compared to HPV-negative HNSCC patients, HPV-positive HNSCC patients were mostly diagnosed with stage IV tumours (29.5 and 66.7 % respectively) (Table 1). This is primarily due to the higher frequency of patients with N2 neck disease that is commonly seen in HPV-positive HNSCC [42]. Most HPV-negative and HPV-positive HNSCC patients were current (31.8 and 22.2 % respectively) and former (45.5 and 51.1 % respectively) smokers (Table 1).
Evaluate the stability of bisulfite-converted DNA
To achieve the uniformity across all of the MSP assays carried out at different times, the stability of the bisulfite converted DNA was tested. MSPs were carried out using converted DNA on five methylated DNA tumour-suppressor genes on a weekly basis for three months. Our densitometry results showed consistency (coefficient of variation, CV of <5 %) across the three month time period when bisulfite converted DNA templates were stored at 4 °C, demonstrating the stability of the MSP reactions (data not shown). All of the MSP data used in this paper were generated within three months’ time period.
Evaluate the specificity of MSP primers
MSP primers for individual tumour-suppressor gene were investigated using bisulfite unconverted DNA. When using bisulphite unconverted DNA, we were unable to detect any PCR amplifications further confirming the specificity of our MSP primers. In addition, as stated above, all of the five DNA methylation tumour genes investigated in this study have been extensively validated previously [6, 25, 33–35].
Evaluate the reproducibility of MSP
Inter and intra-assay variations were carried out using randomised samples for all five methylated DNA tumour-suppressor genes. The inter- and intra-assay CVs fell within the range of 10 to 20 % for all of the studied genes. The limit of detection for our MSP assays were: 1.25 ng/μL of bisulfite-converted DNA for RASSF1α, 20 ng/μL of bisulfite-converted DNA for p16INK4a and TIMP3 and 25 ng/μL of bisulfite-converted DNA for PCQAP respectively.
Five individual tumour-suppressor gene DNA methylation levels in saliva collected from HNSCC patients and normal healthy controls
The five individual tumour-suppressor gene DNA methylation levels showed no significant association with age. DNA methylation levels were relatively higher in saliva collected from HPV-negative patients whilst lower in saliva collected from HPV-positive HNSCC patients compared with normal healthy controls (Additional file 4: Table S2). RASSF1α, PCQAP 5′ and PCQAP 3′ were significantly (p < 0.0001, p < 0.0001 and p < 0.005 respectively) hypermethylated in saliva collected from HPV-negative HNSCC patients whilst p16INK4a, PCQAP 5′and PCQAP 3′ were significantly (p < 0.005, p < 0.05 and p < 0.005 respectively) hypomethylated in the saliva collected from HPV-positive HNSCC patients compared with normal healthy controls (Fig. 2). Table 3 summarises the predictive accuracies for the five individual tumour-suppressor genes.

Overall DNA methylation profiles in the three groups. Whisker-box plot for the methylation signatures of a RASSF1α, b p16INK4a, c TIMP3, d PCQAP 5′ and e PCQAP 3′ in the saliva of normal healthy controls (n = 122), HPV-positive (n = 45) and HPV-negative (n = 88) HNSCC patients with inter-quartile range and median shown using non-parametric Mann-Whitney’s U-test. Significant difference between each categories were marked with * = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001, respectively
Differential diagnosis of HPV-negative and HPV-positive HNSCC patients using the five tumour-suppressor gene panel
The Carstensen’s multivariate receiving operating characteristic, ROC curve offers the best case scenario of the panel’s performance based on the original samples that were used in building the model (Fig. 3). With this approach, this panel performed extremely well with the area under curve (AUC) of 0.86, sensitivity of 71 % and specificity of 80 % when discriminating HPV-negative HNSCC patients from normal healthy controls; and AUC of 0.80, sensitivity of 80 % and specificity of 74 % when comparing HPV-positive HNSCC patients with normal healthy controls (Fig. 3). The data was then processed using five-fold cross-validation and bootstrap to determine the performance of this panel in a ‘most likely scenario’ with the intention of clinical translation. The results obtained suggest that the panel is more appropriate for HPV-negative HNSCC diagnosis as the sensitivity and specificity were least influenced by the enforced probability (Table 4).

Performance of the panel in detecting HPV-negative and positive HNSCC. Carstensen’s multivariate receiver-operating characteristics curve when all of the five salivary methylation genes are combined, comparing normal healthy controls (n = 122) with HPV-negative HNSCC patients (n = 88) (blue bar); and normal healthy controls (n = 122) with HPV-positive (n = 45) HNSCC patients (red bar) respectively
TCGA data for HNSCC tumour and normal tissues
The criteria for probes selection for individual tumour-suppressor gene are based on whether the probes are situated in the region of methylated cites amplified by MSPs. Four of our DNA methylation loci could be found in the TCGA data base and these were RASSF1α, TIMP3, PCQAP 5′ and PCQAP 3′. We were unable to locate a corresponding probe relevant to p16INK4a in the TCGA data base (Additional file 5: Figure S3). While the methylation data for RASSF1α from TCGA correlated with the DNA methylation levels in saliva collected from HPV-positive HNSCC patients, the overall methylation status of TIMP3 and PCQAP did not vary significantly in tumour samples compared to salivary DNA methylation levels. This may be due to the differences in anatomical sites where tumours have been analysed in the TCGA data.
Discussion
Differential DNA methylation in tumour-suppressor genes is a frequent event during human neoplasms [43]. DNA methylation plays a significant role in head and neck carcinogenesis [26, 27, 44]. In this study, we describe a five DNA methylation panel (RASSF1α, p16INK4a, TIMP3, PCQAP 5′ and PCQAP 3′) that can discriminate HPV-negative and HPV-positive HNSCC patients from normal healthy control smokers and non-smokers. Significantly higher DNA methylation levels were observed in saliva collected from HPV-negative HNSCC patients compared with normal healthy controls. In contrast, a significant reduction in DNA methylation was detected in saliva collected from HPV-positive HNSCC patients compared with HPV-negative HNSCC patients. In general, DNA methylation levels were similar or lower in the saliva collected from HPV-positive HNSCC patients compared with the saliva collected from normal healthy controls. Our data corroborates previously published findings that HPV integration reduces global methylation levels [45, 46].
DNA methylation in tumour-suppressor genes is an early event in tumorigenesis; hence, it is likely to represent an ideal biomarker to evaluate early-stage tumour activities [43]. Based on the current literature, all four of the genes analysed in our study have vital roles in regulating cell proliferation either directly or indirectly [47–56]. Down regulation of RASSF1α was found in many cancer types including head and neck, lung, breast, prostate, ovarian, gastric, bladder and colorectal [57–64]. Promoter regions of RASSF1α were found to be hypermethylated in tumour tissues compared to normal tissues [57–63]. In addition, numerous studies have shown that the DNA methylation levels in saliva for RASSF1α mirrors actual tumour activities [6, 26, 34, 65].
p16INK4a protein expression in tumour tissue samples is a current gold stand to determine HPV status in HNSCC patients [32, 66]. This is due to the fact that while the promoter region of p16INK4a is hypermethylated in most cancer types, it was found to be significantly hypomethylated (elevated protein expression) in HPV-positive HNSCC tumour tissues as well as in saliva samples [67]. During HPV-16 integration, HPV-16 E7 binds to pRb and releases E2F which then result in rapid cellular proliferation, resulting in higher expression of p16INK4a [68]. A significant reduction in p16INK4a DNA methylation was observed in saliva from HPV-positive HNSCC patients compared with saliva from normal healthy controls, further confirming the diagnostic utility of p16INK4a protein expression in tumour tissues for determining HPV status. In addition, our findings also corroborated with previous literature, further enforcing the distinct biological and clinical features between HPV-positive and HPV-negative HNSCC patients [5, 7, 69].
TIMP3 DNA methylation levels were higher in saliva collected from HPV-negative HNSCC patients compared to normal healthy controls. DNA promoter hypermethylation of TIMP3 has shown to be strongly associated with HNSCC pathogenesis [70–72]. According to recent publications, DNA methylation of TIMP3 is a robust biomarker, which can also predict HNSCC recurrences [34, 70–73]. In addition, the TIMP3 methylation levels in tumour tissues were able to predict the formation of secondary tumours [73].
While RASSF1α, p16INK4a and TIMP3 have been extensively studied as useful biomarkers to detect HNSCC, PCQAP/MED15 has been identified by our group [25]. In this study, we were able to demonstrate unequivocally that the salivary DNA methylation levels of PCQAP/MED15 could discriminate between normal healthy controls and HPV-negative and HPV-positive HNSCC patients. PCQAP/MED15 encodes for a protein complex member of the transcriptional co-activator mediator family, specifically the RNA polymerase II transcriptional subunit 15 [51]. It is responsible for the transcriptional regulation of ligand-activated proteins such as the transforming growth factor betas (TGFβs) [51]. TGFβs play a role in cellular regulation, proliferation and differentiation [51]. As such, PCQAP/MED15 may have an important role as a tumour-suppressor gene [51]. According to PubMeth database (Ghent University, Ghent, Kortrijk, Belgium), PCQAP/MED15 is hypermethylated in over 66 % of oesophageal and 40 % of prostate cancers. This gene contains two annotated CpG islands, one at the main promoter region and another overlapping with the 14th exon (Additional file 3: Figure S2).
Standard MSP is often regarded as a qualitative analysis; it is also not informative when determining the percentage of methylation levels [74]. However, to quantify methylation bands, we used MSP coupled with densitometry software such as ImageJ. We have also made sure that the band intensities were not saturated and that all of the MSPs were in the linear range of the calibration standard. Based on the results, the MSP assay for the five individual tumour-suppressor genes has been optimised to operate in a linear range (R2 > 0.8). In order to minimise inter-assay variation, two independent researchers quantified the bands and an average value was taken when the results deviated >10 %. The CV of our assays fell within the range of acceptable precision and repeatability, demonstrating that the results are reliable. In addition, the efficiency of MSP was also investigated to address nested-MSP bias.
Conclusion
The differential DNA methylation in tumour-suppressor genes can potentially be used in identifying early-stage HPV-negative HNSCC patients as well as to classify their HPV status accordingly. This indicates that not only can this panel recognize but also categorize patients based on their salivary DNA methylation signature profiles. We’ve also used two advanced statistical methods to demonstrate the clinical relevance of this panel. Our panel was subjected to a five-fold cross-validation and bootstrap statistical analyses and was able to detect HPV-negative HNSCC with high sensitivity and specificity. This is a great clinical end point as it would mean that testing a single saliva sample with a simple DNA methylation test; one would be able to accurately discern three clinical outcomes for a patient in a non-invasive fashion. Furthermore, since the DNA is isolated from saliva, tumour-suppressor methylation signature changes are likely to have originated from tumour cells. In the future, randomised, multi-site and double-blinded studies will be a highly-informative prelude to clinical implementation of this panel to detect and discriminates HPV-positive and HPV-negative HNSCC.
Abbreviations
- AUC:
-
Area under curve
- HNSCC:
-
Head and neck squamous cell carcinoma
- HPV:
-
Human papillomavirus
- IHC:
-
Immunohistochemistry
- MSP:
-
Methylation-specific polymerase chain reaction
- PAH:
-
Princess Alexandra Hospital
- ROC:
-
Receiver operating characteristic
- TCGA:
-
The Cancer Genome Atlas
- TGFβ:
-
Transforming growth factor beta
- TNM:
-
Tumour-node-metastasis
References
Argiris A, et al. Head and neck cancer. Lancet. 2008;371(9625):1695–709.
Lim Y, et al. Salivary epigenetic biomarkers in head and neck squamous cell carcinomas. Biomark Med. 2016;10(3):301–13.
Chang MC, et al. Cell-mediated immunity and head and neck cancer: with special emphasis on betel quid chewing habit. Oral Oncol. 2005;41(8):757–75.
Walden MJ, Aygun N. Head and neck cancer. Semin Roentgenol. 2013;48(1):75–86.
Pfister DG, Fury MG. New chapter in our understanding of human papillomavirus-related head and neck cancer. J Clin Oncol. 2014;32(30):3349–52.
Ovchinnikov DA, et al. Tumor-suppressor gene promoter hypermethylation in saliva of head and neck cancer patients. Transl Oncol. 2012;5(5):321–6.
Syrjanen S. The role of human papillomavirus infection in head and neck cancers. Ann Oncol. 2010;21 Suppl 7:vii243–5.
Chaturvedi AK, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29(32):4294–301.
Gillison ML, et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J Natl Cancer Inst. 2008;100(6):407–20.
Ragin CC, Modugno F, Gollin SM. The epidemiology and risk factors of head and neck cancer: a focus on human papillomavirus. J Dent Res. 2007;86(2):104–14.
Lin H-S, et al. Autoantibody approach for serum-based detection of head and neck cancer. Cancer Epidemiol Biomarkers Prev. 2007;16(11):2396–405.
Worsham MJ. Identifying the risk factors for late stage head and neck cancer. Expert Rev Anticancer Ther. 2011;11(9):1321–5.
Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45(4–5):309–16.
Hall SF, et al. TNM-based stage groupings in head and neck cancer: application in cancer of the hypopharynx. Head Neck. 2009;31(1):1–8.
Price KA, Cohen EE. Current treatment options for metastatic head and neck cancer. Curr Treat Options Oncol. 2012;13(1):35–46.
Villa A, Villa C, Abati S. Oral cancer and oral erythroplakia: an update and implication for clinicians. Aust Dent J. 2011;56(3):253–6.
Mehanna H, et al. Head and neck cancer—Part 1: epidemiology, presentation, and prevention. BMJ. 2010;341:c4684.
Iorgulescu G. Saliva between normal and pathological. Important factors in determining systemic and oral health. J Med Life. 2009;2(3):303–7.
Pfaffe T, et al. Diagnostic potential of saliva: current state and future applications. Clin Chem. 2011;57(5):675–87.
Malamud D. Saliva as a diagnostic fluid. Dent Clin North Am. 2011;55(1):159–78.
na. Head and neck cancer biomarkers detected in saliva. Cancer Biol Ther. 2004. 4(1):6–12.
Mohamed R, et al. The impact of saliva collection and processing methods on CRP, IgE, and Myoglobin immunoassays. Clin Transl Med. 2012;1:19.
Topkas E, et al. Evaluation of saliva collection devices for the analysis of proteins. Clin Chim Acta. 2012;413(13–14):1066–70.
Zhang X, Dimeski G, Punyadeera C. Validation of an immunoassay to measure plasminogen-activator inhibitor-1 concentrations in human saliva. Biochem Med. 2014;24(2):258–65.
Ovchinnikov DA, et al. DNA methylation at the novel CpG sites in the promoter of MED15/PCQAP gene as a biomarker for head and neck cancers. Biomark Insights. 2014;9:53–60.
Demokan S, Dalay N. Role of DNA methylation in head and neck cancer. Clin Epigenetics. 2011;2(2):123–50.
Worsham MJ, et al. Delineating an epigenetic continuum in head and neck cancer. Cancer Lett. 2014;342(2):178–84.
Carvalho AL, et al. Detection of promoter hypermethylation in salivary rinses as a biomarker for head and neck squamous cell carcinoma surveillance. Clin Cancer Res. 2011;17(14):4782–9.
Herman JG, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93(18):9821–6.
Tollefsbol TO, et al. Epigenetics protocols. New York: Humana Press; 2011.
Huang Z, Bassil C, Murphy S. Methylation-specific PCR. In: Malek A, Tchernitsa O, editors. Ovarian cancer. New York: Humana Press; 2013. p. 75–82.
Chai RC, et al. A pilot study to compare the detection of HPV-16 biomarkers in salivary oral rinses with tumour p16INK4a expression in head and neck squamous cell carcinoma patients. BMC Cancer. 2016;16(1):1–8.
Divine KK, et al. Nested multigene MSP/DHPLC method for analyzing promoter hypermethylation status in clinical samples. BioTechniques. 2006. 40(1):40, 42, 44 passim.
Righini CA, et al. Tumor-specific methylation in saliva: a promising biomarker for early detection of head and neck cancer recurrence. Clin Cancer Res. 2007;13(4):1179–85.
Eads CA, et al. Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res. 2001;61(8):3410–8.
Jiang L, et al. CAL 27 is an oral adenosquamous carcinoma cell line. Oral Oncol. 2009;45(11):e204–7.
Carstensen B. Function to compute and draw ROC-curves. 2013. http://bendixcarstensen.com/Epi/
Kohavi R. A study of cross-validation and bootstrap for accuracy estimation and model selection. In: Proceedings of the 14th international joint conference on Artificial intelligence, vol. 2. Montreal: Morgan Kaufmann Publishers Inc; 1995. p. 1137–43.
Geisser S. Predictive inference. New York: Chapman and Hall; 1993.
Devijver PA. Pattern recognition: a statistical approach. London: Prentice-Hall; 1982.
Tang K-W, et al. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun. 2013;4:2513.
Ang KK, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363(1):24–35.
Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–59.
Arantes LM, et al. Methylation as a biomarker for head and neck cancer. Oral Oncol. 2014;50(6):587–92.
Richards KL, et al. Genome-wide hypomethylation in head and neck cancer is more pronounced in HPV-negative tumors and is associated with genomic instability. PLoS One. 2009;4(3):e4941.
Poage GM, et al. Global hypomethylation identifies loci targeted for hypermethylation in head and neck cancer. Clin Cancer Res. 2011;17(11):3579–89.
Dong SM, et al. Epigenetic inactivation of RASSF1A in head and neck cancer. Clin Cancer Res. 2003;9(10):3635–40.
Demokan S, et al. Promoter methylation and loss of p16(INK4a) gene expression in head and neck cancer. Head Neck. 2012;34(10):1470–5.
Guan Z, et al. Promoter methylation and expression of TIMP3 gene in gastric cancer. Diagn Pathol. 2013;8(1):1–6.
Shinojima T, et al. Heterogeneous epigenetic regulation of TIMP3 in prostate cancer. Epigenetics. 2012;7(11):1279–89.
Sandhu HK, et al. An association study of PCQAP polymorphisms and schizophrenia. Psychiatr Genet. 2004;14(3):169–72.
Imielinski M, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–20.
Zhao M, et al. Mediator MED15 modulates transforming growth factor beta (TGFbeta)/Smad signaling and breast cancer cell metastasis. J Mol Cell Biol. 2013;5(1):57–60.
Guo SX, et al. Hypermethylation of p16 and p15 genes and RB protein expression in acute leukemia. Leuk Res. 2000;24(1):39–46.
Calmon MF, et al. Methylation profile of genes CDKN2A (p14 and p16), DAPK1, CDH1, and ADAM23 in head and neck cancer. Cancer Genet Cytogenet. 2007;173(1):31–7.
Sanchez-Cespedes M, et al. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000;60(4):892–5.
Pan J, et al. Association between RASSF1A promoter methylation and prostate cancer: a systematic review and meta-analysis. PLoS One. 2013;8(9):e75283.
Meng W, et al. Combined RASSF1A and RASSF2A promoter methylation analysis as diagnostic biomarker for bladder cancer. Mol Biol Int. 2012;2012:8.
Shi H, et al. Association between RASSF1A promoter methylation and ovarian cancer: a meta-analysis. PLoS One. 2013;8(10):e76787.
Xu J, et al. Methylation of HIN-1, RASSF1A, RIL and CDH13 in breast cancer is associated with clinical characteristics, but only RASSF1A methylation is associated with outcome. BMC Cancer. 2012;12:243.
Shi DT, et al. Association of RASSF1A promoter methylation with gastric cancer risk: a meta-analysis. Tumour Biol. 2014;35(2):943–8.
Wang HL, et al. Aberrant promoter methylation of RASSF1A gene may be correlated with colorectal carcinogenesis: a meta-analysis. Mol Biol Rep. 2014;41(6):3991–9.
Gao T, et al. The association of RAS Association Domain Family Protein1A (RASSF1A) methylation states and bladder cancer risk: a systematic review and meta-analysis. PLoS One. 2012;7(11):e48300.
Han S, et al. Epidemiology and cost analysis for patients with oral cancer in a university hospital in China. BMC Public Health. 2010;10:196.
Koutsimpelas D, et al. Promoter methylation of MGMT, MLH1 and RASSF1A tumor suppressor genes in head and neck squamous cell carcinoma: pharmacological genome demethylation reduces proliferation of head and neck squamous carcinoma cells. Oncol Rep. 2012;27(4):1135–41.
Chai RC, et al. Current trends in the etiology and diagnosis of HPV-related head and neck cancers. Cancer Med. 2015;4(4):596–607.
Konig F, et al. Relation between human papillomavirus positivity and p16 expression in head and neck carcinomas--a tissue microarray study. Anticancer Res. 2007;27(1a):283–8.
Nakao Y, et al. Induction of p16 during immortalization by HPV 16 and 18 and not during malignant transformation. Br J Cancer. 1997;75(10):1410–6.
Boscolo-Rizzo P, et al. New insights into human papillomavirus-associated head and neck squamous cell carcinoma. Acta Otorhinolaryngol Ital. 2013;33(2):77–87.
De Schutter H, et al. Promoter methylation of TIMP3 and CDH1 predicts better outcome in head and neck squamous cell carcinoma treated by radiotherapy only. Oncol Rep. 2009;21(2):507–13.
Rettori MM, et al. Prognostic significance of TIMP3 hypermethylation in post-treatment salivary rinse from head and neck squamous cell carcinoma patients. Carcinogenesis. 2013;34(1):20–7.
Rettori MM, et al. TIMP3 and CCNA1 hypermethylation in HNSCC is associated with an increased incidence of second primary tumors. J Transl Med. 2013;11:316.
Sun W, et al. Detection of TIMP3 promoter hypermethylation in salivary rinse as an independent predictor of local recurrence-free survival in head and neck cancer. Clin Cancer Res. 2012;18(4):1082–91.
Swift-Scanlan T, et al. Two-color quantitative multiplex methylation-specific PCR. Biotechniques. 2006;40(2):210–9.
Acknowledgements
We thank Professor William B. Coman for his clinical input in this study. We also thank Ms Dana Middleton and the staff at the ENT Department of the Princess Alexandra Hospital, Woolloongabba, Australia for their assistance in consenting patients for this study.
Funding
This study was supported by the Queensland Centre for Head and Neck Cancer funded by Atlantic Philanthropies, the Queensland Government, and the Princess Alexandra Hospital.
Availability of data and materials
All data and materials supporting the findings of this study can be found in the body of the manuscript as well as in the supplementary section.
Authors’ contributions
YKL collected samples, compiled all clinical and epidemiological data on all patients, conducted methylation assays, data interpretation and wrote the manuscript, YXW contributed to the methylation assays, DV performed statistical analysis, DAO helped to draft the manuscript and illustrated supplementary figure, CFLP identified areas of tumour cellularity for all patients and conducted HPV assays on selected patients, MJD contributed to statistical analysis and TCGA data interpretation, CP participated in study concept and design, study coordination and revised the manuscript critically for content. All authors have read and approved the content of this manuscript.
Competing interest
The authors declare that they have no competing interests.
Consent for publication
Not applicable
Ethics
This study is approved by the University of Queensland’s (HREC no.: 2014000679) and Queensland University of Technology’s (HREC no.: 1400000617) Medical Ethical Institutional Board and the Princess Alexandra Hospital’s (PAH) Ethics Review Board (HREC no.: HREC/12/QPAH/381). All participants were informed consent for this study.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Figure S1.
MED15/PCQAP MSP amplicon sequence confirmation. The alignment of MED15/PCQAP MSP amplicon sequence in the NCBI Basic Local Alignment Search Tool (BLAST) database. (DOCX 218 kb)
Additional file 2: Table S1.
The Cancer Genome Atlas data file. The raw data extracted from The Cancer Genome Atlas (TCGA) database, containing the methylation signature information for RASSF1α, TIMP3 and PCQAP in HNSCC and normal tissues. (XLSX 101 kb)
Additional file 3: Figure S2.
The amplification regions of individual genes with respective promoter start sites and CpG islands. A detailed map illustrating the location of the amplification region, promoter start site and CpG islands for RASSF1α, p16INK4a, TIMP3 and PCQAP. (DOCX 260 kb)
Additional file 4: Table S2.
Methylation ratio data file. The raw data generated for this study. Each table contains the band intensity ratio of methylated to unmethylated MSP. The file contains the methylation ratio of RASSF1α, p16INK4a, TIMP3 and PCQAP for normal healthy controls and HPV-negative and HPV-positive HNSCC patients used in this study. (XLSX 31 kb)
Additional file 5: Figure S3.
Methylation pattern in tumours and normal tissues. The methylation signature of RASSF1α, TIMP3 and PCQAP in HNSCC and normal tissues from The Cancer Genome Atlas (TCGA) database. (DOCX 91 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lim, Y., Wan, Y., Vagenas, D. et al. Salivary DNA methylation panel to diagnose HPV-positive and HPV-negative head and neck cancers. BMC Cancer 16, 749 (2016). https://doi.org/10.1186/s12885-016-2785-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-016-2785-0