
Abstract
Head and neck cancer (HNC) is a heterogenous and complex entity including diverse anatomical sites and a variety of tumor types displaying unique characteristics and different etilogies. Both environmental and genetic factors play a role in the development of the disease, but the underlying mechanism is still far from clear. Previous studies suggest that alterations in the genes acting in cellular signal pathways may contribute to head and neck carcinogenesis. In cancer, DNA methylation patterns display specific aberrations even in the early and precancerous stages and may confer susceptibility to further genetic or epigenetic changes. Silencing of the genes by hypermethylation or induction of oncogenes by promoter hypomethylation are frequent mechanisms in different types of cancer and achieve increasing diagnostic and therapeutic importance since the changes are reversible. Therefore, methylation analysis may provide promising clinical applications, including the development of new biomarkers and prediction of the therapeutic response or prognosis. In this review, we aimed to analyze the available information indicating a role for the epigenetic changes in HNC.
Similar content being viewed by others
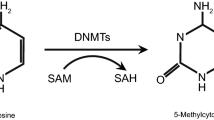

Introduction
Head and neck cancer (HNC) is a highly heterogenous group of malignant diseases and the sixth most frequently observed cancer type in developing countries (Crowe et al. 2002; Ohshima et al. 2005). It reveals different combinations of various sites and disease types which result from sequential genetic changes in multiple intracellular pathways and inherent viral infections. HNC displays serial dysplastic alterations before acquiring invasive characteristics. Tobacco and/or alcohol use are the main etiological factors and play an important role in oral cavity, pharynx, and larynx cancer (Ohshima et al. 2005). Acute laryngitis which can be caused by chronic irritation, inhalation of irritants, viral infections, or allergic reactions may also increase the risk of cancer development (Kumar et al. 2000). The prognosis of the disease varies according to tumor size, local invasion, histology, and grade as well as ethnic origin. These diverse varieties reflect the versatile pathogenesis of the disease. Identification of novel therapeutic targets and new and specific biomarkers for the early detection of HNC could greatly increase the survival rate and might also help as prognostic indicators.
Head and neck carcinogenesis is a multistep and multifactorial complex mechanism containing a variety of genetic and epigenetic abnormalities in DNA repair, signal transduction, apoptosis, angiogenesis, proliferation, differentiation, and cell cycle regulation (Scully et al. 2000). In recent years, the role of epigenetic alterations have been increasingly recognized. Changes in the methylation patterns are one of the most frequent events in human tumors (Jones and Baylin 2002). Two types of methylation changes are usually observed : Hypermethylation of the CpG islands and global hypomethylation in a variety of tumors. DNA hypomethylation has been associated with the activation of oncogenes and chromosomal instability leading to overexpression of the oncogenes, while DNA hypermethylation is associated with the repression of tumor suppressor genes (TSG) and genomic instability. DNA methylation also plays an important role in tumor initiation and progression (Jones and Baylin 2002; Momparler and Bovenzi 2000). Thus, gene silencing by hypermethylation is an important mechanism that has great promise for therapy and for the discovery of new biomarkers. Hovewer, the information on the frequency and specificity of methylation changes is still insufficient. It presents a challenge to identify crucial genes that are susceptible to methylation-induced silencing and are directly associated with the development of HNC. Since HNC is a heterogenous disease, the methylation status may vary according to clinical characteristics and environmental and genetic factors such as anatomic involvement of tumor (Dikshit et al. 2007; Azarschab et al. 2003), human papilloma virus (HPV) positivity (Bennett et al. 2010; Richards et al. 2009), smoking (Sharma et al. 2010), microsatellite instability (MSI) (Zuo et al. 2009; Demokan et al. 2006; Sengupta et al. 2007; Liu et al. 2003a, b, 2002), and geographic location (Li et al. 2003a, b; Ayadi et al. 2008). Therefore, analysis of the epigenetic changes in genes playing important roles in these vital molecular mechanisms is crucial in order to understand the molecular biology of head and neck carcinogenesis.
Genetic evidence indicates that the majority of squamous HNC originate from hyperplastic fields characterized by higher numbers of proliferating cells and clonal selection (Rubin 2011; Tabor et al. 2002). Almost two thirds of the tumor recurrences following resection occur in these regions (Tabor et al. 2004; Ha and Califano 2003). Clonal selection, divergence, and expression are the basic characteristics of the field development. Molecular evidence including cells harboring mutations of the TSG and recent data on p53 mutations support the field cancerization theory. The presence of cells with p53 gene mutations in these fields have also been shown in HNC (Tabor et al. 2001). The genetic changes characterizing these regions can be used to identify individuals at risk of developing cancer (Dakubo et al. 2007). Furthermore, methylation in noncancerous tissues is now considered as a marker for potential cancer risk and previous exposure to certain carcinogenic factors (Nakajima et al. 2008). Knowledge of the methylation status in these regions may enable intervention by using demethylating agents as chemopreventive means (Dakubo et al. 2007). An epigenetic field effect has been first described by increased aberrant methylation in normal tissue from patients with liver cancer (Kondo et al. 2000). Later studies have unequivocally shown the presence of an epigenetic field for cancerization (Ushijima 2007). Recently, the presence of an epigenetic field for cancerization has also been shown in colon (Shen et al. 2005), breast (Yan et al. 2006), and renal (Arai et al. 2006) cancers.
Recent advances in technology make it possible to analyze DNA methylation by highly sensitive and quantitative methods. For DNA methylation analysis, a variety of different methodologies have been used, almost all of which rely on three different approaches for treating DNA: the action of methylation-specific endonucleases, bisulfite modification of unmethylated cytosines, or immunoprecipitation (IP) of the methylated DNA fragments. Combination of these initial steps with different detection techniques for analysis have resulted in various analytical methods to investigate candidate genes or to study global DNA methylation. The main methodologies used for methylation analysis are summarized in Table 1. Several techniques initially confined to smaller regions of the genome have now been extended to perform analysis of the whole genome. New high-throughput methodologies provide information to characterize methylated sequences at single-base resolution on a genome-wide scale.
Recent studies have shown that hypermethylation of some TSG may be a valuable biomarker (Kim et al. 2006; Demokan et al. 2010; Kaur et al. 2010; Pattani et al. 2010) in different cancer types. Furthermore, results of clinical studies using DNA methyltransferase and histone deacetylase inhibitors indicate the potential of epigenetic therapeutics in clinical oncology (Ren et al. 2011; Wagner et al. 2010). The epigenetic changes may help to predict the prognosis and identify individuals who may benefit from the therapy with demethylating agents (Issa et al. 2004).
In this review, we aimed to summarize the present state of knowledge in head and neck carcinogenesis by analyzing the studies investigating the relationship between methylation and HNC. The publications in the literature were retrieved by literature and keyword search using the ISI, PubMed, and Scopus databases to identify the studies relevant to this review. Citations in these publications were also evaluated for their relevance.
Data from analysis of tumor suppressor gene panels
In recent years, most studies have focused on the analysis of promoter methylation of TSG panels playing a role in vital cellular mechanisms via the candidate gene strategy approach (Table 2). In a study among the North American population (Demokan et al. 2010), KIF1A and EDNRB genes were highly methylated (97% and 98%, respectively) in the primary tumor tissue and salivary rinse samples from patients with squamous cell carcinoma, while normal salivary and normal mucosal samples were minimally methylated. A significant association between KIF1A methylation and tumor site was reported. Kaur et al. (2010) found high methylation levels of the KIF1A, EDNRB, DCC, and p16 genes in Indian patients with oral squamous cell carcinoma (OSCC) and Pattani et al. (2010) have shown that, in salivary samples from patients with premalignant or malignant oral cavity lesions, promoter hypermethylation of the KIF1A and EDNRB genes was associated with malignancy. The endothelin receptor type B (EDNRB) is a G protein-coupled receptor which activates a phosphatidylinositol–calcium second messenger system (Smollich and Wülfing 2008). The kinesin family member 1A (KIF1A) gene encodes a protein that is a microtubule-dependent molecular motor involved in important intracellular functions such as organelle transport and cell division (Okada et al. 1995).
We have evaluated the epigenetic changes specific to head and neck squamous cell carcinoma (HNSCC) by investigating promoter hypermethylation of a panel of 24 TSG via candidate gene approach in a recent study (Yalniz et al. 2011). CHFR, RARβ, DAPK1, and RASFF1A genes were found to be the most frequently methylated genes in HNC tumor tissue by methylation-specific multiplex ligation-dependent probe amplification. A further collaborative study analyzing a panel of 22 genes confirmed these findings and found that the RARβ, APC, and CHFR genes were frequently hypermethylated in HNC (Chen et al. 2007).
In another study, six genes were analyzed in patients with HNSCC treated by radiotherapy (de Schutter et al. 2009). The MGMT and TIMP3 genes displayed higher methylation rates. Promoter hypermethylation of the TIMP3 and CDH1 genes were significantly associated with better locoregional control (LRC) and overall survival (OS) or disease-free survival (DFS). TIMP3 methylation highly correlated with DAPK methylation, indicating a very strong functional association between these two genes (Nayak et al. 2007). In other gene panel studies, methylation of the TIMP3, CDH1, p16, MGMT, DAPK, and RASSF1 genes were observed in HNSCC tumors and paired saliva samples (Righini et al. 2007; Hasegawa et al. 2002; Rosas et al. 2001; Sanchez-Cespedes et al. 2000). A study analyzing promoter methylation of 15 candidate genes (Steinmann et al. 2009) in tumors and matched normal tissue from patients with HNSCC has shown that methylation of the p16, MGMT, DAPK, RARβ, hMLH1, CDH1, RASSF2, RASSF5, and MST1 genes was significantly more frequent in the tumors than the normal tissue (Steinmann et al. 2009). The hMLH1 (Puri et al. 2005), RARβ (Maruya et al. 2004), p16, and MGMT (Puri et al. 2005; Maruya et al. 2004) genes have also been found to be frequently methylated in HNSCC. The higher increased methylation of the TSG is usually associated with advanced tumor stages and undifferentiated HNSCC. This has been primarily shown for the p16 and RASSF5 genes (Steinmann et al. 2009). In the same study, RASSF4 gene methylation was more frequent in patients with advanced tumor stage and recurrent HNSCC than patients without relapse. Higher methylation of the p16, RARβ, and RASSF1 genes was also reported in a study in which matched normal samples were not available (Okami et al. 2005).
In a study investigating three TSG functioning in carcinogen metabolism (CYP1A1, CYP2A13, and GSTM1), the genes were found to be moderately (27.4–58.1%) methylated in the tumors, while methylation levels in normal tissue were much lower. A significant correlation was reported between smoking and the methylation status of the CYP1A1 and CYP2A13 genes (Sharma et al. 2010).
Hypermethylation of the hMLH1 gene, which plays an important role in DNA mismatch repair, has been found to be significantly associated with decreased hMLH1 protein expression, MSI, and decreased cause-specific survival for HNSCC patients in various studies (Zuo et al. 2009; Demokan et al. 2006; Sengupta et al. 2007; Liu et al. 2003a, b, 2002). Methylation of both hMLH1 and hMSH2 genes may have an important role in oral carcinogenesis and have been associated with the susceptibility for oral malignancies (Czerninski et al. 2009).
Biomarkers predicting clinical response, tumor recurrence, or patient survival are not available for many cancer types, particularly for oral and pharyngeal cancer. Taioli et al. (2009) have studied the methylation of a panel of TSG in order to identify a possible correlation with survival and recurrence rates in patients with oral or pharyngeal cancer. MGMT promoter methylation was inversely associated with poor OS and DFS, indicating that MGMT promoter methylation may act as a possible prognostic biomarker for oral and pharyngeal cancer (Taioli et al. 2009). In oral epithelial dysplasia that transforms to OSCC, p16 methylation may act as a candidate biomarker of malignant transformation, whereas methylation of the MGMT, CYGB, and CCNA1 genes are not associated with malignancy (Hall et al. 2008). In oral carcinomas, methylation of the p16 and MGMT genes are frequently observed as an early event (Kato et al. 2006). Promoter hypermethylation of the p16, p15, hMLH1, MGMT, and CDH1 genes has been reported in OSCC (Viswanathan et al. 2003). The Ras/PI3K/AKT pathway is a major mechanism associated with radioresistance in OSCC. A study investigating four genes (RASSF1A, RASSF2A, PTEN, and HIN-1) in this pathway revealed that RASSF1A and RASSF2A methylation were more frequent in the tumors and significantly associated with poor DFS (Huang et al. 2009). Methylation of CCNA1, CYGB, and p16 genes has also been correlated with the clinicopathological parameters in oral cancer (Shaw et al. 2006). In OSCC, different levels of methylation have been reported for the p16, p15, p14, DCC, DAPK, MINT1, MINT2, MINT27, and MINT31 gene promoters and DCC methylation was associated with bone invasion of gingival tumors, invasiveness, and reduced survival (Ogi et al. 2002). Aberrant methylation of the p14, p15, and p16 gene promoters has been reported in HNSCC (Weber et al. 2002), oral precancerous lesions (Takeshima et al. 2008), oral carcinomas (Sailasree et al. 2008; Yeh et al. 2003; Shintani et al. 2001), and salivary gland carcinomas (Nishimine et al. 2003). When normal tissue and benign and malignant salivary gland tumors were compared, significantly higher methylation of the APC, RARβ, MINT1, PGP9.5, and TIMP3 genes were observed in salivary duct carcinoma (Durr et al. 2010). In adenoid cystic carcinomas of the salivary gland, p16, RASSF1A, and DAPK gene methylation is also a common event (Li et al. 2005).
In patients with laryngeal and hypopharyngeal cancers, promoter methylation of the p16, MGMT, DAPK, and CDH1 genes are observed frequently (Dikshit et al. 2007; Azarschab et al. 2003). For tumors in the oropharynx, a statistically significant association between hypermethylation of the DAPK1 gene and risk of lymph node metastases has been reported, and significant evidence indicate an association between hypermethylation of the ADAM23 gene and advanced tumor stage in larynx cancer (Calmon et al. 2007).
In a study investigating the methylation status of candidate TSG in surgical margins as a predictor of local recurrence in HNSCC, it has been shown that analysis of the CDKN2A, CCNA1, and DCC genes in the surgical margins by quantitative methylation-specific PCR (QMSP) can correctly predict local recurrences in HNSCC (Tan et al. 2008). Using candidate gene and discovery approaches, 21 genes were investigated in HNSCC and normal tissue samples. p16, MINT31, and RASSF1A gene methylation were detected only in HNSCC but not in the controls (Carvalho et al. 2008).
Data from mRNA expression array studies
Recently, studies investigating novel methylated TSG specific to the tumor types have made use of mRNA expression arrays to analyze more than 40,000 genetic regions on a single platform via pharmacological unmasking and discovery approaches (Yamashita et al. 2002; Tokumaru et al. 2004).
After bisulfite sequencing, rapid subtractive hybridization, and microarray analysis in order to determine genes that are induced to reexpression by the demethylating agent 5-aza-2′-deoxycytidine in HNSCC cell lines, 35 out of 78 genes were selected and only 3 of these (CRABP2, MX1, and SLC15A3) were verified by QMSP (Calmon et al. 2009). After methylation-specific PCR (MSP) analysis, CRABP2 and MX1 genes were highly methylated in primary HNSCC compared with lymphocytes from a healthy cohort. In addition, lack of the CRABP2 protein was associated with poor survival rates, indicating that CRABP2 expression may be a potential prognostic biomarker for patients with HNSCC.
In a study, PGP9.5, CCNA1, bone morphogenetic protein 2A (BMP2A), metallothionein 1G (MT1G), and neuromedin U (NmU) genes were highly methylated and CCNA1 hypermethylation displayed an inverse correlation with p53 mutations (Tokumaru et al. 2004).
Methylation of the Nischarin (NISCH), p21-activated protein kinase 3 (PAK3), KIF1A, and OGDHL genes has been reported in 8–52% of the patients with HNC, whereas no methylation was observed in the normal cohort (Hoque et al. 2008).
Global methylation
A recent study indicated the involvement of global DNA hypermethylation in the pathogenesis of HNSCC (Worsham et al. 2010). Using DNA IP and Affymetrix whole-genome tiling arrays, 231 new and previously unreported genes out of 1,143 cancer genes on the array were identified via the whole-genome methylation approach.
In a study aiming to characterize early molecular changes in premalignant lesions of the oral cavity and the role of tobacco and alcohol consumption or HPV infections, the global methylation index was found to be 4.28 (95%CI, 4.1, 4.4) in the oral cancer case series. Methylation was inversely associated with tobacco use (Guerrero-Preston et al. 2009).
In cancer, abnormal demethylation leads to the loss of silencing in repetitive elements which are located on approximately 50% of the human genome, whereas in normal cells, these repetitive sequences are regulated by epigenetic silencing. DNA methylation alterations in HNC and adjacent nontumor tissues was investigated via a genome-wide microarray approach (Szpakowski et al. 2009). Of the more than 250,000 repetitive elements probed, between 5% and 8% displayed disease-related DNA methylation changes. Among the SVA, HERV, LINE-1P, AluY, and MaLR families, LINE-1 (Richards et al. 2009; Subbalekha et al. 2009; Smith et al. 2007), SINE (Alu) (Richards et al. 2009) repetitive elements, and LRE1 (Furniss et al. 2008; Hsiung et al. 2007) showed loss of DNA methylation in the tumors when compared to matched normal adjacent tissue. LINE and LRE1 hypomethylation were more frequently observed in HPV-negative than in HPV-positive tumors (Richards et al. 2009).
The Runt-related transcription factor 3 (RUNX3) gene plays a role in the transforming growth factor-beta (TGF-β)-induced tumor suppression pathway. Although RUNX3 has been considered as a TSG in some studies (Bae et al. 1995; Li et al. 2002), it has been shown that the expression level of RUNX3 in HNSCC tissues are higher than that in normal oral epithelial tissues due to demethylation (Ginos et al. 2004; Salto-Tellez et al. 2006). Therefore, it has been suggested that RUNX3 may have an oncogenic role in HNSCC and its expression may predict malignant behavior and the effect of chemotherapeutic drugs in HNSCC as a potential biomarker (Tsunematsu et al. 2009). In contrast to HNSCC, the RUNX3 gene is underexpressed in OSCC due to promoter hypermethylation, indicating that RUNX3 plays an important role in oral carcinogenesis and may be a useful diagnostic marker and a potential therapeutic target for OSCC (Gao et al. 2009).
A recent report has shown that the SEPT9, SLC5A8, FUSSEL18, EBF3, and IRX1 genes which act in the TGF-β signaling pathway are commonly methylated and downregulated in HNC (Bennett et al. 2009). In IP studies, all these candidate genes were observed to interact with the components of the TGF-β pathway (Bennett et al. 2008). It was reported that decreased mitotic activity and increased apoptosis rates were observed when the SLC5A8, EBF3, and IRX1 genes are overexpressed.
Recently, HPV-infected HNSCC tumors have been shown to display higher levels of global DNA methylation. These tumors were universally methylated irrespective of the clinical factors and methylation of the FUSSEL18, IRX1, and EBF3 genes were likely corralated with recurrences. Promoter methylation of the FUSSEL18 and SEPTIN9 genes was significantly associated with alcohol and tobacco consumption. A trend between HPV16 positivity and methylation of the IRX1, EBF3, SLC5A8, and SEPT9 genes was noted and it has been suggested that this gene panel may be used for the selection of treatment modality (Bennett et al. 2010).
The Transketolase-like 1 (TKTL1) gene is a novel candidate oncogene, which is hypomethylated in human HNSCC tumor samples and contributes to HNSCC carcinogenesis via aerobic glycolysis and HIF1-α stabilization (Sun et al. 2010).
Studies of individual genes
Tumor suppressor genes
Significantly higher methylation of the p14ARF gene has been reported in OSCC compared to normal control tissues, implying that the methylation status of p14ARF may be an important determinant in the early diagnosis and treatment of OSCC (Kordi-Tamandani et al. 2010; Ishida et al. 2005).
Data on the methylation of the p16 gene promoter is not consistent. The gene has been analyzed individually or in gene panels but with discordant results. In a study, p16 methylation has been associated with malignant transformation of oral epithelial dysplasia and was considered a potential biomarker for the prediction of prognosis of mild or moderate oral epithelial dysplasia (Cao et al. 2009; Kresty et al. 2002). High methylation of the p16 promoter region has also been reported in carcinomas of the tongue and methylation in the surgical margins were found to increase the risk of local recurrences 6.3-fold when compared with patients with negative margins (Sinha et al. 2009). Varying degrees of p16 methylation has been reported in OSCC (Ohta et al. 2009; Ruesga et al. 2007; Nakahara et al. 2006; Huang et al. 2002; Yakushiji et al. 2001; Nakahara et al. 2001; Miracca et al. 1999; Riese et al. 1999; El-Naggar et al. 1997; Tao et al. 1997; González et al. 1997; Reed et al. 1996), in mucoepidermoid carcinoma (MEC) of the salivary glands (Guo et al. 2007; Agnese et al. 2006), in HNC (Yalniz et al. 2011; Koscielny et al. 2007; Ai et al. 2003), and in larynx cancer (Smigiel et al. 2004). Conversely, there is a also a report of low p16 methylation in larynx cancer (Agnese et al. 2006).
DCC is a candidate TSG located on chromosome 18q21. Hypermethylation of DCC as a mechanism for inactivation in HNSCC has been investigated (Carvalho et al. 2006). The DCC promoter was highly methylated in the tumors and there was a significant correlation between DCC promoter region hypermethylation and lack of DCC expression.
In a study investigating the methylation and expression levels of the APC and SFRP genes, both genes were highly methylated in MEC but not in adjacent normal tissue. There was significant correlation between methylation and low SFRP1 expression. Methylation of the SFRP1 gene was the main cause of decreased SFRP1 expression. Beta-catenin expression was also associated with reduced SFRP1 expression. In addition, both SFRP1 and beta-catenin expression were associated with tumor grade and stage. Survival was particularly poor in patients with reduced SFRP1 and cytoplasmic/nuclear beta-catenin expression. It has been suggested that detection of SFRP1 expression and aberrant beta-catenin expression in the cell may be useful biomarkers of tumor progression and prognosis in patients with MEC (Lee et al. 2010).
Alterations in TGF-β signaling are common in HNSCC. Hypermethylation of TGF-β type I receptor (TGFBR-I) gene was evaluated via MSP and restriction enzyme-mediated PCR (MSRE). TGFBR-I expression was lost in 83% of the HNSCC tumors and was linked to DNA hypermethylation of the CpG-rich promoter region in 62% of the samples (Muñoz-Antonia et al. 2009).
NDRG2 is a candidate TSG involved in oral squamous cell cancers via the Akt signaling pathway. Reduced NDRG2 mRNA levels, caused by promoter methylation, have been reported in most of the OSCC patients and in several cases of precancerous leukoplakia with dysplasia (Furuta et al. 2010).
Hypermethylation of the FancB gene has been observed in sporadic HNSCC tumors (Smith et al. 2010), while methylation of the SYK gene has been frequently observed in OSCC; the downregulation of SYK expression due to promoter methylation was associated with metastasis (Ogane et al. 2009). MGMT gene methylation is observed in laryngeal cancer (Zhang et al. 2006, 2004) and HNSCC (Zuo et al. 2004), but not in normal larynx tissue.
The MALT1 gene, responseible for activating nuclear factor-kappaB in lymphocyte lineages, is located in a genomic region encoding putative TSG and is expressed in the nucleus of oral epithelial cells. Absence of expression due to epigenetic inactivation during tumor progression has been associated with tumor recurrence and poor patient survival, suggesting that analysis of MALT1 expression may be a useful predictive and prognostic marker for OSCC (Chiba et al. 2009).
In primary OSCC, aberrant methylation of the RASSF2 gene and a high frequency of ROBO1 methylation have been reported in early dysplastic lesions of the head and neck (Imai et al. 2008; Ghosh et al. 2009). A significant inverse correlation between RASSF1A promoter methylation and HPV infection in HNSCC has been shown (Dong et al. 2003).
GALR1 methylation is observed in primary HNSCC tumors and correlates with decreased GALR1 expression, as well as increased tumor size, lymph node status, tumor stage, CCND1 expression, p16 methylation, and survival (Misawa et al. 2008).
Methylation of the CDH1 gene promoter has been reported in tongue cancer (Chang et al. 2002), salivary gland adenoid cystic carcinoma (Zhang et al. 2007a), and nonmetastatic oral cancer as an early event (De Moraes et al. 2008; Maeda et al. 2007a; Yeh et al. 2002).
Expression of the RARβ gene has been studied in primary tissue specimens of different anatomical sites from patients with HNSCC and a strong correlation was found between hypermethylation and reduced expression of RARβ2 (Youssef et al. 2004). In particular, significantly lower hypermethylation and higher RARβ2 mRNA expression levels when compared to the tumors located at other sites of the head and neck were observed in tumors from the hypopharynx (Olasz et al. 2007) and salivary duct or acinic cell carcinomas displaying RASSF1 methylation (Williams et al. 2006).
Genes with different functions
CHFR is a putative early mitotic checkpoint gene which causes a delay in chromosome condensation in response to mitotic stress. In a study, aberrant promoter methylation of the CHFR gene was reported in patients with OSCC, while the gene was methylated minimally in the surrounding normal mucosa and no methylation was observed in the CHFR promoter in a healthy cohort (Baba et al. 2009; Toyota et al. 2003).
The HIC1 gene plays a role in the regulation of transcription. A study using MSP has reported that HIC1 is highly (95%) methylated in HNSCC and reexpression of the gene was associated with decreased aggressiveness (Brieger et al. 2010). On the other hand, in a recent study from our group, no significant methylation of the HIC1 gene was observed in patients with HNC using a more sensitive technique (Yalniz et al. 2011).
The CYGB gene was first described as an intracellular globin of unknown function (Burmester et al. 2002). CYGB downregulation is a key event in the familial cancer syndrome of the upper aerodigestive tract. Increased expression of the CYGB gene displays an inverse correlation with promoter methylation and a strong correlation with tumor hypoxia. It has been consistently associated with aggressive tumors in oral and oropharyngeal squamous cell carcinoma when compared with histologically tumor-free surgical margins (Shaw et al. 2009).
In individual studies, aberrant methylation of RB1 (Kishi et al. 2005) and 14-3-3 sigma (Uchida et al. 2004; Gasco et al. 2002) in salivary gland cancer, Apaf-1 (Huang et al. 2004) and DAPK (Zhang and Kong 2004) in laryngeal squamous cell carcinoma, TSC2 (Chakraborty et al. 2008), SFRP1, SFRP2, SFRP5 (Sogabe et al. 2008), RECK (Long et al. 2008), EpCAM (Shiah et al. 2009), MTNR1A (Nakamura et al. 2008), IKKalpha (Maeda et al. 2007b), PRTFDC1 (Suzuki et al. 2007), LRP1B (Nakagawa et al. 2006), DBCCR1 (Gao et al. 2004), and 14-3-3 sigma (Gasco et al. 2002) in OSCC, PGP9.5 (Tokumaru et al. 2008), C/EBPalpha (Bennett et al. 2007), LHX6 (Estécio et al. 2006), STAT1 (Xi et al. 2006), TIG1 (Tokumaru et al. 2005), TCF21 (Smith et al. 2006), SOCS3 (Weber et al. 2005), p15 (Chang et al. 2004), and ATM (Ai et al. 2004) genes in patients with HNC have also been reported.
miR-137 plays an important role in the cell cycle control. It has been shown that miR-137 and miR-193a are epigenetically silenced during oral carcinogenesis (Kozaki et al. 2008). Methylation of miR-137 has been reported in squamous cell carcinoma tissue and oral rinse samples and was associated with gender and inversely associated with body mass index (Langevin et al. 2010).
Specific lysine residues in histone tails are methylated, providing an epigenetic marker that modulates biological functions changing the heterochromatin structure and leading to tumor development. H3K4 histone methylation was investigated in OSCC, dysplastic lesions, and normal tissue samples (Piyathilake et al. 2005). The levels of H3K4me2 and H3K4me3 displayed striking variations in OSCC when compared with normal tissue and leukoplakias. The me2 levels were increased while the me3 levels decreased in the tumors (Mancuso et al. 2009).
Nasopharyngeal carcinoma and studies of TSG gene panels
Nasopharyngeal carcinoma (NPC) is a rare malignancy with unique genetic, viral, and environmental characteristics that distinguish it from other types of head and neck carcinoma. It has a different etiology, epidemiology, prognosis, and therapy. The clinical management of NPC remains challenging largely due to the lack of early detection strategies for this tumor (Loyo et al. 2011; Razak et al. 2010).
A recent study investigating a panel of 18 marker genes in nasopharyngeal tumors has shown that the methylation status of the AIM1, APC, CALCA, DCC, DLEC, DLC1, ESR, FHIT, KIF1A, and PGP9.5 genes were significantly associated with NPC when compared with other tumors or the benign nasopharyngeal biopsy samples (Loyo et al. 2011).
In an 11-gene panel, promoter methylation levels of CDH1, p15, THBS1, RASSF1A, MLH1, MGMT, p16, and TP73 genes were significantly higher in the tumor samples from patients with NPC when compared with the lymphocytes from the same individuals (Wong et al. 2003a).
Using the discovery approach after expression profiling among eight potential candidate TSG, promoter methylation of only three genes (CCNA1, RARRES1, and HRASLS) have been significantly associated with NPC (Yanatatsaneejit et al. 2008).
Promoter methylation of the RASSF1A, DAPK, and RARβ2 genes was analyzed by MSP in primary NPC tumors and normal nasopharyngeal epithelia. All genes were highly methylated in tumor tissue, whereas methylation was not observed in the normal nasopharyngeal tissue. Methylation of the three genes was significantly associated with lymph node involvement. RASSF1A and RARβ2 methylation also correlated with age at diagnosis, T stage, and histological type (Fendri et al. 2009). Epigenetic silencing of cellular retinol-binding proteins, CRBPI, CRBPIV, and RARβ2 are also commonly observed in NPC tumor samples (Kwong et al. 2005a, 2002).
High methylation frequencies for the DAPK, RASSF1A, CDH1, and p16 genes are frequently observed in NPC tumors (Kwong et al. 2002; Chang et al. 2003a). In contrast to HNSCC (Dikshit et al. 2007), in NPC, no change has been reported in the RUNX3 promoter region, whereas the p16, RASSF1A, CDH1, and hMLH1 gene promoters were frequently methylated (Tan et al. 2006).
In the patients with NPC from Southeast Asia but not from North Africa, methylation of p16, DLEC1, BLU, and CDH1 genes were found to be associated with the juvenile and adult forms of the disease. Strong correlations were observed between aberrant promoter methylation of the CDH1 and BLU genes and lymph node invasion (Li et al. 2003a, b) or undifferentiated tumors, respectively (Ayadi et al. 2008).
Single-gene studies in NPC
Tumor suppressor genes
WIF1 is a highly conserved gene on chromosome 12 and encodes a protein of the sFRP family, which inhibits the Wnt signaling pathway (Kawano and Kypta 2003). Inhibition of Wnt signaling induces apoptosis and inhibits tumor growth in many cancer types (He et al. 2004). Silencing of the WIF1 gene by hypermethylation may result in the activation of some tumors (Suzuki et al. 2004; Caldwell et al. 2004; Ai et al. 2006; Taniguchi et al. 2005). The WIF1 promoter region is highly methylated in nasopharyngeal tumors, whereas no methylation is observed in the normal mucosa (Fendri et al. 2010; Chan et al. 2007). WIF1 methylation has been found to be associated with tumor size, node involvement and metastasis, and age (Fendri et al. 2010).
Expression of the MIPOL1 gene is also downregulated in some NPC tumors via promoter hypermethylation and allelic loss (Cheung et al. 2009). Likewise, DAB2 is frequently methylated in NPC, which correlates with the loss of expression in NPC tumors (Tong et al. 2010).
Two studies investigating the RASSF1A gene have shown that the gene is highly methylated in primary NPC but not in normal nasopharyngeal epithelia (Wang et al. 2009; Lo et al. 2001). It has been suggested that aberrant hypermethylation of RASSF1A and high Epstein–Barr virus (EBV) load may play an important role in NPC carcinogenesis (Zhou et al. 2005).
Methylation of CDH1 is more frequently observed in advanced stages of NPC (Niemhom et al. 2008). Hypermethylation of CDH1 promoter and presence of EBV are predominantly detected in undifferentiated and nonkeratinizing NPC compared to squamous cell NPC. Most of the NPC samples demonstrating CDH1 hypermethylation were EBV-positive, whereas the EBV genome and hypermethylation were not detected in normal nasopharyngeal tissue when CDH1 meythylation was absent, indicating a correlation between CDH1 hypermethylation and EBV infection (Niemhom et al. 2008; Krishna et al. 2005; Li et al. 2003a, b; Tsao et al. 2003; Kao et al. 2002).
Genes with various funcitons
14-3-3 sigma, the downstream target of p53, is a negative regulator of cell cycle G2–M phase checkpoint in response to DNA damage. By MSP, 100% methylation of 14-3-3 sigma was shown in tumor tissue but not in any of adjacent normal nasopharyngeal epithelial tissue (Yi et al. 2009). Analysis by real-time PCR, Western blotting, and immunohistochemistry have revealed that 14-3-3 sigma expression is downregulated or absent in NPC samples displaying high methylation. In addition, hypermethylation of 14-3-3 sigma has been associated with lymph node and distant metastasis.
Methylation of the PGP9.5 gene is frequently detected in primary NPC but only minimally observed in normal nasopharyngeal tissue, indicating that the methylation-mediated silencing of PGP9.5 may be important in nasopharyngeal carcinogenesis (Li et al. 2010). The EDNRB gene, located on chromosome 13q22, was highly methylated in primary NPC tumor samples, while no methylation was observed in normal nasopharyngeal epithelia (Zhou et al. 2007; Lo et al. 2002). In patients with NPC, high methylation of the LARS2 gene, which is located at the chromosome 3 common eliminated region-1 (C3CER1) on 3p21.3, has been reported (Zhou et al. 2009).
Several genes which have been studied in gene panels were also evaluated in single-gene studies. RASFF2 (Zhang et al. 2007b), DLC1 (Seng et al. 2007), DAPK (Kong et al. 2006; Wong et al. 2002), CHFR (Cheung et al. 2005), TIG1 (Kwong et al. 2005b), BLU (Qiu et al. 2004; Liu et al. 2003a, b), and p16 (Lo et al. 1996) are among those. The corresponding data are given in Table 3. Other newly identified TSG are IRF8 (Lee et al. 2008), ADAMTS18, which is a novel gene located on 16q23 (Jin et al. 2007), LTF (Yi et al. 2006), CDH13 (Sun et al. 2007), PCDH10 (Ying et al. 2006), TSLC1 (Hui et al. 2003), HIN-1 (Wong et al. 2003b), and RIZ1 (Chang et al. 2003b) The GNAT1 gene has been shown to be methylated not only in all primary NPC tissues but also in 80% of tissue samples with chronic nasopharyngitis (Yi et al. 2007).
Conclusions
Biomarkers predicting clinical response, tumor recurrence, or patient survival are not available for HNC. Further studies are needed to identify new biomarkers for early detection and prediction of the therapeutic response or prognosis. Follow-up studies using quantitative MSP analysis, global methylation profiling, and detailed analysis of downstream DNA repair genes regulated by promoter methylation may provide new insight into the issue. Considering the great heterogeneity of HNC, a combination of multiple genes for analysis may provide a higher coverage for diverse tumors than the analysis of a single gene (Carvalho et al. 2008). Recent data indicate that promoter hypermethylation of the KIF1A and EDNRB genes is a frequent event in primary HNSCC and combining only the KIF1A and EDNRB genes provides a higher specificity and sensitivity than using a panel of 10 different genes (Demokan et al. 2010). Preferential methylation of these genes in salivary rinses from HNSCC patients may provide a promising potential biomarker for the disease. Other genes investigated frequently and found to display significant differential methylation are RASSF1A, DAPK1, MGMT, RARβ, CDH1, hMLH1, and CHFR, although a wide range of different methylation ratios have been reported. These variations most probably result from the differences in the sensitivity of the methods, variations in the processing of the samples, and composition of the patient cohorts.
Methylation analysis has the additional advantage that methylation patterns are not affected by external factors or temporary physiological changes. They persist and are usually increased during disease progression. The higher specificity and sensitivity provides a suitable tool to obtain predictive or prognostic information. So far, the information about DNA methylation has not translated into useful and reliable markers for HNC in the clinical practice. However, given the sensitive and high-throughput quantitative methodologies for methylation analysis, specific markers for HNC will certainly emerge by careful evaluation and combination of different marker panels to achieve a high sensitivity for the disease. Over the next years, clinical tests based on diagnostic and therapeutic methylation markers will certainly be available and will be used for the assessment of prognosis, treatment planning, and to predict the response.
Future studies aiming to determine the causative role and significance of these epigenetic alterations may provide important clues into the mechanism and contribution of the specific events and help to establish a sequence of methylation events during tumor development associated with different stages of head and neck carcinogenesis. Since tumor-specific DNA can be easily detected in blood, serum, and saliva, methylation analysis can be used as an noninvasive method for the early detection using a panel of the genes specific for the disease. Furthermore, epigenetic silencing of the genes offers new therapeutic approaches using demethylating agents. Analysis of genome-wide methylation profiles by new high-throughput technologies which enable simultaneous analysis of thousands of genetic loci will certainly help to identify highly specific novel methylation biomarkers.
References
Agnese V, Corsale S, Calò V, Augello C, Bruno L, Calcara D, Crosta A, Rodolico V, Rinaldi G, Cicero G, Latteri F, Agrusa A, Morello V, Adamo V, Altavilla G, Di Fede G, Fiorentino E, Grassi N, Latteri MA, Valerio MR, Tomasino RM, Colucci G, Bazan V, Russo A, Gruppo Oncologico dell'Italia Meridionale (2006) Significance of p16INK4A hypermethylation gene in primary head/neck and colorectal tumors: it is a specific tissue event? Results of a 3-year GOIM (Gruppo Oncologico dell'Italia Meridionale) prospective study. Ann Oncol 17(Suppl 7):137–141
Ai L, Stephenson KK, Ling W, Zuo C, Mukunyadzi P, Suen JY, Hanna E, Fan CY (2003) The p16 (CDKN2a/INK4a) tumor-suppressor gene in head and neck squamous cell carcinoma: a promoter methylation and protein expression study in 100 cases. Mod Pathol 16:944–950
Ai L, Vo QN, Zuo C, Li L, Ling W, Suen JY, Hanna E, Brown KD, Fan CY (2004) Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: promoter hypermethylation with clinical correlation in 100 cases. Cancer Epidemiol Biomarkers Prev 13:150–156
Ai L, Tao Q, Zhong S, Fields CR, Kim WJ, Lee MW, Cui Y, Brown KD, Robertson KD (2006) Inactivation of Wnt inhibitory factor-1 (WIF1) expression by epigenetic silencing is a common event in breast cancer. Carcinogenesis 27:1341–1348
Arai E, Kanai Y, Ushijima S, Fujimoto H, Mukai K, Hirohashi S (2006) Regional DNA hypermethylation and DNA methyltransferase (DNMT) 1 protein overexpression in both renal tumors and corresponding nontumorous renal tissues. Int J Cancer 119:288–296
Ayadi W, Karray-Hakim H, Khabir A, Feki L, Charfi S, Boudawara T, Ghorbel A, Daoud J, Frikha M, Busson P, Hammami A (2008) Aberrant methylation of p16, DLEC1, BLU and E-cadherin gene promoters in nasopharyngeal carcinoma biopsies from Tunisian patients. Anticancer Res 28:2161–2167
Azarschab P, Stembalska A, Loncar MB, Pfister M, Sasiadek MM, Blin N (2003) Epigenetic control of E-cadherin (CDH1) by CpG methylation in metastasising laryngeal cancer. Oncol Rep 10:501–503
Baba S, Hara A, Kato K, Long NK, Hatano Y, Kimura M, Okano Y, Yamada Y, Shibata T (2009) Aberrant promoter hypermethylation of the CHFR gene in oral squamous cell carcinomas. Oncol Rep 22:1173–1179
Bae SC, Takahashi E, Zhang YW, Ogawa E, Shigesada K, Namba Y, Satake M, Ito Y (1995) Cloning, mapping and expression of PEBP2aC, a third gene encoding the mammalian runt domain. Gene 159:245–248
Bennett KL, Hackanson B, Smith LT, Morrison CD, Lang JC, Schuller DE, Weber F, Eng C, Plass C (2007) Tumor suppressor activity of CCAAT/enhancer binding protein alpha is epigenetically down-regulated in head and neck squamous cell carcinoma. Cancer Res 67:4657–4664
Bennett KL, Karpenko M, Lin MT, Claus R, Arab K, Dyckhoff G, Plinkert P, Herpel E, Smiraglia D, Plass C (2008) Frequently methylated tumor suppressor genes in head and neck squamous cell carcinoma. Cancer Res 68:4494–4499
Bennett KL, Romigh T, Eng C (2009) Disruption of transforming growth factor-beta signaling by five frequently methylated genes leads to head and neck squamous cell carcinoma pathogenesis. Cancer Res 69:9301–9305
Bennett KL, Lee W, Lamarre E, Zhang X, Seth R, Scharpf J, Hunt J, Eng C (2010) HPV status-independent association of alcohol and tobacco exposure or prior radiation therapy with promoter methylation of FUSSEL18, EBF3, IRX1, and SEPT9, but not SLC5A8, in head and neck squamous cell carcinomas. Genes Chromosomes Cancer 49:319–326
Brieger J, Pongsapich W, Mann SA, Hedrich J, Fruth K, Pogozelski B, Mann WJ (2010) Demethylation treatment restores hic1 expression and impairs aggressiveness of head and neck squamous cell carcinoma. Oral Oncol 46:678–683
Burmester T, Ebner B, Weich B, Hankeln T (2002) Cytoglobin: a novel globin type ubiquitously expressed in vertebrate tissues. Mol Biol Evol 19:416–421
Caldwell GM, Jones C, Gensberg K, Jan S, Hardy RG, Byrd P, Chughtai S, Wallis Y, Matthews GM, Morton DG (2004) The Wnt antagonist sFRP1 in colorectal tumorigenesis. Cancer Res 64:883–888
Calmon MF, Colombo J, Carvalho F, Souza FP, Filho JF, Fukuyama EE, Camargo AA, Caballero OL, Tajara EH, Cordeiro JA, Rahal P (2007) Methylation profile of genes CDKN2A (p14 and p16), DAPK1, CDH1, and ADAM23 in head and neck cancer. Cancer Genet Cytogenet 173:31–37
Calmon MF, Rodrigues RV, Kaneto CM, Moura RP, Silva SD, Mota LD, Pinheiro DG, Torres C, de Carvalho AF, Cury PM, Nunes FD, Nishimoto IN, Soares FA, da Silva AM, Kowalski LP, Brentani H, Zanelli CF, Silva WA Jr, Rahal P, Tajara EH, Carraro DM, Camargo AA, Valentini SR (2009) Epigenetic silencing of CRABP2 and MX1 in head and neck tumors. Neoplasia 11:1329–1339
Cao J, Zhou J, Gao Y, Gu L, Meng H, Liu H, Deng D (2009) Methylation of p16 CpG island associated with malignant progression of oral epithelial dysplasia: a prospective cohort study. Clin Cancer Res 15:5178–5183
Carvalho AL, Chuang A, Jiang WW, Lee J, Begum S, Poeta L, Zhao M, Jerónimo C, Henrique R, Nayak CS, Park HL, Brait MR, Liu C, Zhou S, Koch W, Fazio VM, Ratovitski E, Trink B, Westra W, Sidransky D, Moon CS, Califano JA (2006) Deleted in colorectal cancer is a putative conditional tumor-suppressor gene inactivated by promoter hypermethylation in head and neck squamous cell carcinoma. Cancer Res 66:9401–9407
Carvalho AL, Jeronimo C, Kim MM, Henrique R, Zhang Z, Hoque MO, Chang S, Brait M, Nayak CS, Jiang WW, Claybourne Q, Tokumaru Y, Lee J, Goldenberg D, Garrett-Mayer E, Goodman S, Moon CS, Koch W, Westra WH, Sidransky D, Califano JA (2008) Evaluation of promoter hypermethylation detection in body fluids as a screening/diagnosis tool for head and neck squamous cell carcinoma. Clin Cancer Res 14:97–107
Chakraborty S, Mohiyuddin SM, Gopinath KS, Kumar A (2008) Involvement of TSC genes and differential expression of other members of the mTOR signaling pathway in oral squamous cell carcinoma. BMC Cancer 8:163
Chan SL, Cui Y, van Hasselt A, Li H, Srivastava G, Jin H, Ng KM, Wang Y, Lee KY, Tsao GS, Zhong S, Robertson KD, Rha SY, Chan AT, Tao Q (2007) The tumor suppressor Wnt inhibitory factor 1 is frequently methylated in nasopharyngeal and esophageal carcinomas. Lab Invest 87:644–650
Chang HW, Chow V, Lam KY, Wei WI, Yuen A (2002) Loss of E-cadherin expression resulting from promoter hypermethylation in oral tongue carcinoma and its prognostic significance. Cancer 94:386–392
Chang HW, Chan A, Kwong DL, Wei WI, Sham JS, Yuen AP (2003a) Detection of hypermethylated RIZ1 gene in primary tumor, mouth, and throat rinsing fluid, nasopharyngeal swab, and peripheral blood of nasopharyngeal carcinoma patient. Clin Cancer Res 9:1033–1038
Chang HW, Chan A, Kwong DL, Wei WI, Sham JS, Yuen AP (2003b) Evaluation of hypermethylated tumor suppressor genes as tumor markers in mouth and throat rinsing fluid, nasopharyngeal swab and peripheral blood of nasopharygeal carcinoma patient. Int J Cancer 105:851–855
Chang HW, Ling GS, Wei WI, Yuen AP (2004) Smoking and drinking can induce p15 methylation in the upper aerodigestive tract of healthy individuals and patients with head and neck squamous cell carcinoma. Cancer 101:125–132
Chen K, Sawhney R, Khan M, Benninger MS, Hou Z, Sethi S, Stephen JK, Worsham MJ (2007) Methylation of multiple genes as diagnostic and therapeutic markers in primary head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg 133:1131–1138
Cheung HW, Ching YP, Nicholls JM, Ling MT, Wong YC, Hui N, Cheung A, Tsao SW, Wang Q, Yeun PW, Lo KW, Jin DY, Wang X (2005) Epigenetic inactivation of CHFR in nasopharyngeal carcinoma through promoter methylation. Mol Carcinog 43:237–245
Cheung AK, Lung HL, Ko JM, Cheng Y, Stanbridge EJ, Zabarovsky ER, Nicholls JM, Chua D, Tsao SW, Guan XY, Lung ML (2009) Chromosome 14 transfer and functional studies identify a candidate tumor suppressor gene, mirror image polydactyly 1, in nasopharyngeal carcinoma. Proc Natl Acad Sci USA 106:14478–14483
Chiba T, Maeda G, Kawashiri S, Kato K, Imai K (2009) Epigenetic loss of mucosa-associated lymphoid tissue 1 expression in patients with oral carcinomas. Cancer Res 69:7216–7223
Crowe DL, Hacia JG, Hsieh CL, Sinha UK, Rice H (2002) Molecular pathology of head and neck cancer. Histol Histopathol 17:909–914
Czerninski R, Krichevsky S, Ashhab Y, Gazit D, Patel V, Ben-Yehuda D (2009) Promoter hypermethylation of mismatch repair genes, hMLH1 and hMSH2 in oral squamous cell carcinoma. Oral Dis 15:206–213
Dakubo GD, Jakupciak JP, Birch-Machin MA, Parr RL (2007) Clinical implications and utility of field cancerization. Cancer Cell Int 7:2
De Moraes RV, Oliveira DT, Landman G, de Carvalho F, Caballero O, Nonogaki S, Nishimoto I, Kowalski LP (2008) E-cadherin abnormalities resulting from CpG methylation promoter in metastatic and nonmetastatic oral cancer. Head Neck 30:85–92
De Schutter H, Geeraerts H, Verbeken E, Nuyts S (2009) Promoter methylation of TIMP3 and CDH1 predicts better outcome in head and neck squamous cell carcinoma treated by radiotherapy only. Oncol Rep 21:507–513
Demokan S, Suoglu Y, Demir D, Gozeler M, Dalay N (2006) Microsatellite instability and methylation of the DNA mismatch repair genes in head and neck cancer. Ann Oncol 17:995–999
Demokan S, Chang X, Chuang A, Mydlarz WK, Kaur J, Huang P, Khan Z, Khan T, Ostrow KL, Brait M, Hoque MO, Liegeois NJ, Sidransky D, Koch W, Califano JA (2010) KIF1A and EDNRB are differentially methylated in primary HNSCC and salivary rinses. Int J Cancer 127:2351–2359
Dikshit RP, Gillio-Tos A, Brennan P, De Marco L, Fiano V, Martinez-Peñuela JM, Boffetta P, Merletti F (2007) Hypermethylation, risk factors, clinical characteristics, and survival in 235 patients with laryngeal and hypopharyngeal cancers. Cancer 110:1745–1751
Dong SM, Sun DI, Benoit NE, Kuzmin I, Lerman MI, Sidransky D (2003) Epigenetic inactivation of RASSF1A in head and neck cancer. Clin Cancer Res 9:3635–3640
Durr ML, Mydlarz WK, Shao C, Zahurak ML, Chuang AY, Hoque MO, Westra WH, Liegeois NJ, Califano JA, Sidransky D, Ha PK (2010) Quantitative methylation profiles for multiple tumor suppressor gene promoters in salivary gland tumors. PLoS One 5:e10828
El-Naggar AK, Lai S, Clayman G, Lee JK, Luna MA, Goepfert H, Batsakis JG (1997) Methylation, a major mechanism of p16/CDKN2 gene inactivation in head and neck squamous carcinoma. Am J Pathol 151:1767–1774
Estécio MR, Youssef EM, Rahal P, Fukuyama EE, Góis-Filho JF, Maniglia JV, Goloni-Bertollo EM, Issa JP, Tajara EH (2006) LHX6 is a sensitive methylation marker in head and neck carcinomas. Oncogene 25:5018–5026
Fendri A, Masmoudi A, Khabir A, Sellami-Boudawara T, Daoud J, Frikha M, Ghorbel A, Gargouri A, Mokdad-Gargouri R (2009) Inactivation of RASSF1A, RARbeta2 and DAP-kinase by promoter methylation correlates with lymph node metastasis in nasopharyngeal carcinoma. Cancer Biol Ther 8:444–451
Fendri A, Khabir A, Hadri-Guiga B, Sellami-Boudawara T, Daoud J, Frikha M, Ghorbel A, Gargouri A, Mokdad-Gargouri R (2010) Epigenetic alteration of the Wnt inhibitory factor-1 promoter is common and occurs in advanced stage of Tunisian nasopharyngeal carcinoma. Cancer Invest 28:896–903
Furniss CS, Marsit CJ, Houseman EA, Eddy K, Kelsey KT (2008) Line region hypomethylation is associated with lifestyle and differs by human papillomavirus status in head and neck squamous cell carcinomas. Cancer Epidemiol Biomarkers Prev 17:966–971
Furuta H, Kondo Y, Nakahata S, Hamasaki M, Sakoda S, Morishita K (2010) NDRG2 is a candidate tumor-suppressor for oral squamous-cell carcinoma. Biochem Biophys Res Commun 391:1785–1791
Gao S, Worm J, Guldberg P, Eiberg H, Krogdahl A, Sørensen JA, Liu CJ, Reibel J, Dabelsteen E (2004) Loss of heterozygosity at 9q33 and hypermethylation of the DBCCR1 gene in oral squamous cell carcinoma. Br J Cancer 91:760–764
Gao F, Huang C, Lin M, Wang Z, Shen J, Zhang H, Jiang L, Chen Q (2009) Frequent inactivation of RUNX3 by promoter hypermethylation and protein mislocalization in oral squamous cell carcinomas. J Cancer Res Clin Oncol 135:739–747
Gasco M, Bell AK, Heath V, Sullivan A, Smith P, Hiller L, Yulug I, Numico G, Merlano M, Farrell PJ, Tavassoli M, Gusterson B, Crook T (2002) Epigenetic inactivation of 14-3-3 sigma in oral carcinoma: association with p16(INK4a) silencing and human papillomavirus negativity. Cancer Res 62:2072–2076
Ghosh S, Ghosh A, Maiti GP, Alam N, Roy A, Roychoudhury S, Panda CK (2009) Alterations of ROBO1/DUTT1 and ROBO2 loci in early dysplastic lesions of head and neck: clinical and prognostic implications. Hum Genet 125:189–198
Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL, Gaffney PM (2004) Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res 64:55–63
González MV, Pello MF, López-Larrea C, Suárez C, Menéndez MJ, Coto E (1997) Deletion and methylation of the tumour suppressor gene p16/CDKN2 in primary head and neck squamous cell carcinoma. J Clin Pathol 50:509–512
Guerrero-Preston R, Báez A, Blanco A, Berdasco M, Fraga M, Esteller M (2009) Global DNA methylation: a common early event in oral cancer cases with exposure to environmental carcinogens or viral agents. P R Health Sci J 28:24–29
Guo XL, Sun SZ, Wang WX, Wei FC, Yu HB, Ma BL (2007) Alterations of p16INK4a tumour suppressor gene in mucoepidermoid carcinoma of the salivary glands. Int J Oral Maxillofac Surg 36:350–353
Ha PK, Califano JA (2003) The molecular biology of mucosal field cancerization of the head and neck. Crit Rev Oral Biol Med 14:363–369
Hall GL, Shaw RJ, Field EA, Rogers SN, Sutton DN, Woolgar JA, Lowe D, Liloglou T, Field JK, Risk JM (2008) p16 Promoter methylation is a potential predictor of malignant transformation in oral epithelial dysplasia. Cancer Epidemiol Biomarkers Prev 17:2174–2179
Hasegawa M, Nelson HH, Peters E, Ringstrom E, Posner M, Kelsey KT (2002) Patterns of gene promoter methylation in squamous cell cancer of the head and neck. Oncogene 21:4231–4236
He B, You L, Uematsu K, Xu Z, Lee AY, Matsangou M, McCormick F, Jablons DM (2004) A monoclonal antibody against Wnt-1 induces apoptosis in human cancer cells. Neoplasia 6:7–14
Hoque MO, Kim MS, Ostrow KL, Liu J, Wisman GB, Park HL, Poeta ML, Jeronimo C, Henrique R, Lendvai A, Schuuring E, Begum S, Rosenbaum E, Ongenaert M, Yamashita K, Califano J, Westra W, van der Zee AG, Van Criekinge W, Sidransky D (2008) Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res 68:2661–2670
Hsiung DT, Marsit CJ, Houseman EA, Eddy K, Furniss CS, McClean MD, Kelsey KT (2007) Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 16:108–114
Huang MJ, Yeh KT, Shih HC, Wang YF, Lin TH, Chang JY, Shih MC, Chang JG (2002) The correlation between CpG methylation and protein expression of P16 in oral squamous cell carcinomas. Int J Mol Med 10:551–554
Huang DF, Fu WN, Shang C, Xu ZM, Li ZG, Sun KL (2004) Expression and promoter methylation of Apaf-1 gene in laryngeal squamous cell carcinoma. Yi Chuan Xue Bao 31:1327–1331
Huang KH, Huang SF, Chen IH, Liao CT, Wang HM, Hsieh LL (2009) Methylation of RASSF1A, RASSF2A, and HIN-1 is associated with poor outcome after radiotherapy, but not surgery, in oral squamous cell carcinoma. Clin Cancer Res 15:4174–4180
Hui AB, Lo KW, Kwong J, Lam EC, Chan SY, Chow LS, Chan AS, Teo PM, Huang DP (2003) Epigenetic inactivation of TSLC1 gene in nasopharyngeal carcinoma. Mol Carcinog 38:170–178
Imai T, Toyota M, Suzuki H, Akino K, Ogi K, Sogabe Y, Kashima L, Maruyama R, Nojima M, Mita H, Sasaki Y, Itoh F, Imai K, Shinomura Y, Hiratsuka H, Tokino T (2008) Epigenetic inactivation of RASSF2 in oral squamous cell carcinoma. Cancer Sci 99:958–966
Ishida E, Nakamura M, Ikuta M, Shimada K, Matsuyoshi S, Kirita T, Konishi N (2005) Promotor hypermethylation of p14ARF is a key alteration for progression of oral squamous cell carcinoma. Oral Oncol 41:614–622
Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, Bayar E, Lyons J, Rosenfeld CS, Cortes J, Kantarjian HM (2004) Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 103:1635–1640
Jin H, Wang X, Ying J, Wong AH, Li H, Lee KY, Srivastava G, Chan AT, Yeo W, Ma BB, Putti TC, Lung ML, Shen ZY, Xu LY, Langford C, Tao Q (2007) Epigenetic identification of ADAMTS18 as a novel 16q23.1 tumor suppressor frequently silenced in esophageal, nasopharyngeal and multiple other carcinomas. Oncogene 26:7490–7498
Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3:415–428
Kao RH, Huang LC, Hsu YH (2002) Mapping the methylation pattern by bisulfite genomic sequencing of the E-cadherin promoter CpG island in nasopharyngeal carcinoma. Anticancer Res 22:4109–4113
Kato K, Hara A, Kuno T, Mori H, Yamashita T, Toida M, Shibata T (2006) Aberrant promoter hypermethylation of p16 and MGMT genes in oral squamous cell carcinomas and the surrounding normal mucosa. J Cancer Res Clin Oncol 132:735–743
Kaur J, Demokan S, Tripathi SC, Macha MA, Begum S, Califano JA, Ralhan R (2010) Promoter hypermethylation in Indian primary oral squamous cell carcinoma. Int J Cancer 127:2367–2373
Kawano Y, Kypta Y (2003) Secreted antagonists of the Wnt signalling pathway. J Cell Sci 116:2627–2634
Kim JS, Kim JW, Han J, Shim YM, Park J, Kim DH (2006) Cohypermethylation of p16 and FHIT promoters as a prognostic factor of recurrence in surgically resected stage I non-small cell lung cancer. Cancer Res 66:4049–4054
Kishi M, Nakamura M, Nishimine M, Ikuta M, Kirita T, Konishi N (2005) Genetic and epigenetic alteration profiles for multiple genes in salivary gland carcinomas. Oral Oncol 41:161–169
Kondo Y, Kanai Y, Sakamoto M, Mizokami M, Ueda R, Hirohashi S (2000) Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis—a comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology 32:970–979
Kong WJ, Zhang S, Guo CK, Wang YJ, Chen X, Zhang SL, Zhang D, Liu Z, Kong W (2006) Effect of methylation-associated silencing of the death-associated protein kinase gene on nasopharyngeal carcinoma. Anticancer Drugs 17:251–259
Kordi-Tamandani DM, Moazeni-Roodi A, Rigi Ladez MA, Hashemi M, Birjandian E, Torkamanzehi A (2010) Analysis of methylation patterns and expression profiles of p14ARF gene in patients with oral squamous cell carcinoma. Int J Biol Markers 25:99–103
Koscielny S, Dahse R, Ernst G, von Eggeling F (2007) The prognostic relevance of p16 inactivation in head and neck cancer. ORL J Otorhinolaryngol Relat Spec 69:30–36
Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J (2008) Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res 68:2094–2105
Kresty LA, Mallery SR, Knobloch TJ, Song H, Lloyd M, Casto BC, Weghorst CM (2002) Alterations of p16(INK4a) and p14(ARF) in patients with severe oral epithelial dysplasia. Cancer Res 62:5295–5300
Krishna SM, Kattoor J, Balaram P (2005) Down regulation of adhesion protein E-cadherin in Epstein–Barr virus infected nasopharyngeal carcinomas. Cancer Biomark 1:271–277
Kumar V, Cotran R, Robbins SL (2000) Basic Pathology, 6th edn. WBSC, Philadelphia
Kwong J, Lo KW, To KF, Teo PM, Johnson PJ, Huang DP (2002) Promoter hypermethylation of multiple genes in nasopharyngeal carcinoma. Clin Cancer Res 8:131–137
Kwong J, Lo KW, Chow LS, Chan FL, To KF, Huang DP (2005a) Silencing of the retinoid response gene TIG1 by promoter hypermethylation in nasopharyngeal carcinoma. Int J Cancer 113:386–392
Kwong J, Lo KW, Chow LS, To KF, Choy KW, Chan FL, Mok SC, Huang DP (2005b) Epigenetic silencing of cellular retinol-binding proteins in nasopharyngeal carcinoma. Neoplasia 7:67–74
Langevin SM, Stone RA, Bunker CH, Grandis JR, Sobol RW, Taioli E (2010) MicroRNA-137 promoter methylation in oral rinses from patients with squamous cell carcinoma of the head and neck is associated with gender and body mass index. Carcinogenesis 31:864–870
Lee KY, Geng H, Ng KM, Yu J, van Hasselt A, Cao Y, Zeng YX, Wong AH, Wang X, Ying J, Srivastava G, Lung ML, Wang LD, Kwok TT, Levi BZ, Chan AT, Sung JJ, Tao Q (2008) Epigenetic disruption of interferon-gamma response through silencing the tumor suppressor interferon regulatory factor 8 in nasopharyngeal, esophageal and multiple other carcinomas. Oncogene 27:5267–5276
Lee CH, Hung YJ, Lin CY, Hung PH, Hung HW, Shieh YS (2010) Loss of SFRP1 expression is associated with aberrant beta-catenin distribution and tumor progression in mucoepidermoid carcinoma of salivary glands. Ann Surg Oncol 17:2237–2246
Li QL, Ito K, Sakakura C, Fukamachi H, Inoue K, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, Kim HM, Kim WJ, Yamamoto H, Yamashita N, Yano T, Ikeda T, Itohara S, Inazawa J, Abe T, Hagiwara A, Yamagishi H, Ooe A, Kaneda A, Sugimura T, Ushijima T, Bae SC, Ito Y (2002) Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 109:113–124
Li Z, Lin SX, Liang YJ (2003a) Detection of gene promoter methylation and mRNA, protein expression levels of E-cadherin in nasopharyngeal carcinoma. Zhonghua Bing Li Xue Za Zhi 32:25–30
Li Z, Lin SX, Liang YJ (2003b) Influence of E-cadherin promoter methylation and mutation of beta-catenin on invasion and metastasis of nasopharyngeal carcinoma cells. Zhonghua Zhong Liu Za Zhi 25:238–242
Li J, El-Naggar A, Mao L (2005) Promoter methylation of p16INK4a, RASSF1A, and DAPK is frequent in salivary adenoid cystic carcinoma. Cancer 104:771–776
Li L, Tao Q, Jin H, van Hasselt A, Poon FF, Wang X, Zeng MS, Jia WH, Zeng YX, Chan AT, Cao Y (2010) The tumor suppressor UCHL1 forms a complex with p53/MDM2/ARF to promote p53 signaling and is frequently silenced in nasopharyngeal carcinoma. Clin Cancer Res 16:2949–2958
Liu K, Huang H, Mukunyadzi P, Suen JY, Hanna E, Fan CY (2002) Promoter hypermethylation: an important epigenetic mechanism for hMLH1 gene inactivation in head and neck squamous cell carcinoma. Otolaryngol Head Neck Surg 126:548–553
Liu K, Zuo C, Luo QK, Suen JY, Hanna E, Fan CY (2003a) Promoter hypermethylation and inactivation of hMLH1, a DNA mismatch repair gene, in head and neck squamous cell carcinoma. Diagn Mol Pathol 12:50–56
Liu XQ, Chen HK, Zhang XS, Pan ZG, Li A, Feng QS, Long QX, Wang XZ, Zeng YX (2003b) Alterations of BLU, a candidate tumor suppressor gene on chromosome 3p21.3, in human nasopharyngeal carcinoma. Int J Cancer 106:60–65
Lo KW, Cheung ST, Leung SF, van Hasselt A, Tsang YS, Mak KF, Chung YF, Woo JK, Lee JC, Huang DP (1996) Hypermethylation of the p16 gene in nasopharyngeal carcinoma. Cancer Res 56:2721–2725
Lo KW, Kwong J, Hui AB, Chan SY, To KF, Chan AS, Chow LS, Teo PM, Johnson PJ, Huang DP (2001) High frequency of promoter hypermethylation of RASSF1A in nasopharyngeal carcinoma. Cancer Res 61:3877–3881
Lo KW, Tsang YS, Kwong J, To KF, Teo PM, Huang DP (2002) Promoter hypermethylation of the EDNRB gene in nasopharyngeal carcinoma. Int J Cancer 98:651–655
Long NK, Kato K, Yamashita T, Makita H, Toida M, Hatakeyama D, Hara A, Mori H, Shibata T (2008) Hypermethylation of the RECK gene predicts poor prognosis in oral squamous cell carcinomas. Oral Oncol 44:1052–1058
Loyo M, Brait M, Kim MS, Ostrow KL, Jie CC, Chuang AY, Califano JA, Liégeois NJ, Begum S, Westra WH, Hoque MO, Tao Q, Sidransky D (2011) A survey of methylated candidate tumor suppressor genes in nasopharyngeal carcinoma. Int J Cancer 128:1393–1403
Maeda G, Chiba T, Aoba T, Imai K (2007a) Epigenetic inactivation of E-cadherin by promoter hypermethylation in oral carcinoma cells. Odontology 95:24–29
Maeda G, Chiba T, Kawashiri S, Satoh T, Imai K (2007b) Epigenetic inactivation of IkappaB Kinase-alpha in oral carcinomas and tumor progression. Clin Cancer Res 13:5041–5047
Mancuso M, Matassa DS, Conte M, Colella G, Rana G, Fucci L, Piscopo M (2009) H3K4 histone methylation in oral squamous cell carcinoma. Acta Biochim Pol 56:405–410
Maruya S, Issa JP, Weber RS, Rosenthal DI, Haviland JC, Lotan R, El-Naggar AK (2004) Differential methylation status of tumor-associated genes in head and neck squamous carcinoma: incidence and potential implications. Clin Cancer Res 10:3825–3830
Miracca EC, Kowalski LP, Nagai MA (1999) High prevalence of p16 genetic alterations in head and neck tumours. Br J Cancer 81:677–683
Misawa K, Ueda Y, Kanazawa T, Misawa Y, Jang I, Brenner JC, Ogawa T, Takebayashi S, Grenman RA, Herman JG, Mineta H, Carey TE (2008) Epigenetic inactivation of galanin receptor 1 in head and neck cancer. Clin Cancer Res 14:7604–7613
Momparler RL, Bovenzi V (2000) DNA methylation and cancer. J Cell Physiol 183:145–154
Muñoz-Antonia T, Torrellas-Ruiz M, Clavell J, Mathews LA, Muro-Cacho CA, Báez A (2009) Aberrant methylation inactivates transforming growth factor Beta receptor I in head and neck squamous cell carcinoma. Int J Otolaryngol 2009:848695
Nakagawa T, Pimkhaokham A, Suzuki E, Omura K, Inazawa J, Imoto I (2006) Genetic or epigenetic silencing of low density lipoprotein receptor-related protein 1B expression in oral squamous cell carcinoma. Cancer Sci 97:1070–1074
Nakahara Y, Shintani S, Mihara M, Ueyama Y, Matsumura T (2001) High frequency of homozygous deletion and methylation of p16(INK4A) gene in oral squamous cell carcinomas. Cancer Lett 163:221–228
Nakahara Y, Shintani S, Mihara M, Hino S, Hamakawa H (2006) Detection of p16 promoter methylation in the serum of oral cancer patients. Int J Oral Maxillofac Surg 35:362–365
Nakajima T, Enomoto S, Ushijima T (2008) DNA methylation: a marker for carcinogen exposure and cancer risk. Environ Health Prev Med 13:8–15
Nakamura E, Kozaki K, Tsuda H, Suzuki E, Pimkhaokham A, Yamamoto G, Irie T, Tachikawa T, Amagasa T, Inazawa J, Imoto I (2008) Frequent silencing of a putative tumor suppressor gene melatonin receptor 1 A (MTNR1A) in oral squamous-cell carcinoma. Cancer Sci 99:1390–1400
Nayak CS, Carvalho AL, Jeronimo C, Henrique R, Kim MM, Hoque MO, Chang S, Jiang WW, Koch W, Westra W, Sidransky D, Califano J (2007) Positive correlation of tissue inhibitor of metalloproteinase-3 and death-associated protein kinase hypermethylation in head and neck squamous cell carcinoma. Laryngoscope 117:1376–1380
Niemhom S, Kitazawa S, Kitazawa R, Maeda S, Leopairat J (2008) Hypermethylation of epithelial-cadherin gene promoter is associated with Epstein–Barr virus in nasopharyngeal carcinoma. Cancer Detect Prev 32:127–134
Nishimine M, Nakamura M, Kishi M, Okamoto M, Shimada K, Ishida E, Kirita T, Konishi N (2003) Alterations of p14ARF and p16INK4a genes in salivary gland carcinomas. Oncol Rep 10:555–560
Ogane S, Onda T, Takano N, Yajima T, Uchiyama T, Shibahara T (2009) Spleen tyrosine kinase as a novel candidate tumor suppressor gene for human oral squamous cell carcinoma. Int J Cancer 124:2651–2657
Ogi K, Toyota M, Ohe-Toyota M, Tanaka N, Noguchi M, Sonoda T, Kohama G, Tokino T (2002) Aberrant methylation of multiple genes and clinicopathological features in oral squamous cell carcinoma. Clin Cancer Res 8:3164–3171
Ohshima H, Tazawa H, Sylla BS, Sawa T (2005) Prevention of human cancer by modulation of chronic imflammatory processes. Mutat Res 591:110–122
Ohta S, Uemura H, Matsui Y, Ishiguro H, Fujinami K, Kondo K, Miyamoto H, Yazawa T, Danenberg K, Danenberg PV, Tohnai I, Kubota Y (2009) Alterations of p16 and p14ARF genes and their 9p21 locus in oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 107:81–91
Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N (1995) The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 81:769–780
Okami K, Sakai A, Onuki J, Hamano T, Iida M, Takahashi M (2005) Promoter hypermethylation of tumor-associated genes in head and neck cancer. Nippon Jibiinkoka Gakkai Kaiho 108:207–213
Olasz J, Juhász A, Remenár E, Engi H, Bak M, Csuka O, Kásler M (2007) RAR beta2 suppression in head and neck squamous cell carcinoma correlates with site, histology and age. Oncol Rep 18:105–112
Pattani KM, Zhang Z, Demokan S, Glazer C, Loyo M, Goodman S, Sidransky D, Bermudez F, Jean-Charles G, McCaffrey T, Padhya T, Phelan J, Spivakovsky S, Bowne HY, Goldberg JD, Rolnitzky L, Robbins M, Kerr AR, Sirois D, Califano JA (2010) Endothelin receptor type B gene promoter hypermethylation in salivary rinses is independently associated with risk of oral cavity cancer and premalignancy. Cancer Prev Res 3:1093–1103
Piyathilake CJ, Bell WC, Jones J, Henao OL, Heimburger DC, Niveleau A, Grizzle WE (2005) Patterns of global DNA and histone methylation appear to be similar in normal, dysplastic and neoplastic oral epithelium of humans. Dis Markers 21:147–151
Puri SK, Si L, Fan CY, Hanna E (2005) Aberrant promoter hypermethylation of multiple genes in head and neck squamous cell carcinoma. Am J Otolaryngol 26:12–17
Qiu GH, Tan LK, Loh KS, Lim CY, Srivastava G, Tsai ST, Tsao SW, Tao Q (2004) The candidate tumor suppressor gene BLU, located at the commonly deleted region 3p21.3, is an E2F-regulated, stress-responsive gene and inactivated by both epigenetic and genetic mechanisms in nasopharyngeal carcinoma. Oncogene 23:4793–4806
Razak AR, Siu LL, Liu FF, Ito E, O'Sullivan B, Chan K (2010) Nasopharyngeal carcinoma: the next challenges. Eur J Cancer 46:1967–1978
Reed AL, Califano J, Cairns P, Westra WH, Jones RM, Koch W, Ahrendt S, Eby Y, Sewell D, Nawroz H, Bartek J, Sidransky D (1996) High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res 56:3630–3633
Ren J, Singh BN, Huang Q, Li Z, Gao Y, Mishra P, Hwa YL, Li J, Dowdy SC, Jiang SW (2011) DNA hypermethylation as a chemotherapy target. Cell Signal 23:1082–1093
Richards KL, Zhang B, Baggerly KA, Colella S, Lang JC, Schuller DE, Krahe R (2009) Genome-wide hypomethylation in head and neck cancer is more pronounced in HPV-negative tumors and is associated with genomic instability. PLoS One 4:e4941
Riese U, Dahse R, Fiedler W, Theuer C, Koscielny S, Ernst G, Beleites E, Claussen U, von Eggeling F (1999) Tumor suppressor gene p16 (CDKN2A) mutation status and promoter inactivation in head and neck cancer. Int J Mol Med 4:61–65
Righini CA, de Fraipont F, Timsit JF, Faure C, Brambilla E, Reyt E, Favrot MC (2007) Tumor-specific methylation in saliva: a promising biomarker for early detection of head and neck cancer recurrence. Clin Cancer Res 13:1179–1185
Rosas SL, Koch W, da Costa Carvalho MG, Wu L, Califano J, Westra W, Jen J, Sidransky D (2001) Promoter hypermethylation patterns of p16, O6-methylguanine-DNA-methyltransferase, and death-associated protein kinase in tumors and saliva of head and neck cancer patients. Cancer Res 61:939–942
Rubin H (2011) Fields and field cancerization: the preneoplastic origins of cancer: asymptomatic hyperplastic fields are precursors of neoplasia, and their progression to tumors can be tracked by saturation density in culture. Bioessays 33:224–231
Ruesga MT, Acha-Sagredo A, Rodríguez MJ, Aguirregaviria JI, Videgain J, Rodríguez C, de Pancorbo ML, Aguirre JM (2007) p16(INK4a) promoter hypermethylation in oral scrapings of oral squamous cell carcinoma risk patients. Cancer Lett 250:140–145
Sailasree R, Abhilash A, Sathyan KM, Nalinakumari KR, Thomas S, Kannan S (2008) Differential roles of p16INK4A and p14ARF genes in prognosis of oral carcinoma. Cancer Epidemiol Biomarkers Prev 17:414–420
Salto-Tellez M, Peh BK, Ito K, Tan SH, Chong PY, Han HC, Tada K, Ong WY, Soong R, Voon DC, Ito Y (2006) RUNX3 protein is overexpressed in human basal cell carcinomas. Oncogene 25:7646–7649
Sanchez-Cespedes M, Esteller M, Wu L, Nawroz-Danish H, Yoo GH, Koch WM, Jen J, Herman JG, Sidransky D (2000) Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res 60:892–895
Scully C, Field JK, Tanzawa H (2000) Genetic aberrations in oral or head and neck squamous cell carcinoma (SCCHN): 1. Carcinogen metabolism, DNA repair and cell cycle control. Oral Oncol 36:256–263
Seng TJ, Low JS, Li H, Cui Y, Goh HK, Wong ML, Srivastava G, Sidransky D, Califano J, Steenbergen RD, Rha SY, Tan J, Hsieh WS, Ambinder RF, Lin X, Chan AT, Tao Q (2007) The major 8p22 tumor suppressor DLC1 is frequently silenced by methylation in both endemic and sporadic nasopharyngeal, esophageal, and cervical carcinomas, and inhibits tumor cell colony formation. Oncogene 26:934–944
Sengupta S, Chakrabarti S, Roy A, Panda CK, Roychoudhury S (2007) Inactivation of human mutL homolog 1 and mutS homolog 2 genes in head and neck squamous cell carcinoma tumors and leukoplakia samples by promoter hypermethylation and its relation with microsatellite instability phenotype. Cancer 109:703–712
Sharma R, Panda NK, Khullar M (2010) Hypermethylation of carcinogen metabolism genes, CYP1A1, CYP2A13 and GSTM1 genes in head and neck cancer. Oral Dis 16:668–673
Shaw RJ, Liloglou T, Rogers SN, Brown JS, Vaughan ED, Lowe D, Field JK, Risk JM (2006) Promoter methylation of P16, RARbeta, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing. Br J Cancer 94:561–568
Shaw RJ, Omar MM, Rokadiya S, Kogera FA, Lowe D, Hall GL, Woolgar JA, Homer J, Liloglou T, Field JK, Risk JM (2009) Cytoglobin is upregulated by tumour hypoxia and silenced by promoter hypermethylation in head and neck cancer. Br J Cancer 101:139–144
Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, Hamilton SR, Issa JP (2005) MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst 97:1330–1338
Shiah SG, Chang LC, Tai KY, Lee GH, Wu CW, Shieh YS (2009) The involvement of promoter methylation and DNA methyltransferase-1 in the regulation of EpCAM expression in oral squamous cell carcinoma. Oral Oncol 45:e1–e8
Shintani S, Nakahara Y, Mihara M, Ueyama Y, Matsumura T (2001) Inactivation of the p14(ARF), p15(INK4B) and p16(INK4A) genes is a frequent event in human oral squamous cell carcinomas. Oral Oncol 37:498–504
Sinha P, Bahadur S, Thakar A, Matta A, Macha M, Ralhan R, Gupta SD (2009) Significance of promoter hypermethylation of p16 gene for margin assessment in carcinoma tongue. Head Neck 31:1423–1430
Smigiel R, Sasiadek M, Krecicki T, Ramsey D, Jagielski J, Blin N (2004) Inactivation of the cyclin-dependent kinase inhibitor 2A (CDKN2A) gene in squamous cell carcinoma of the larynx. Mol Carcinog 39:147–154
Smith LT, Lin M, Brena RM, Lang JC, Schuller DE, Otterson GA, Morrison CD, Smiraglia DJ, Plass C (2006) Epigenetic regulation of the tumor suppressor gene TCF21 on 6q23-q24 in lung and head and neck cancer. Proc Natl Acad Sci USA 103:982–987
Smith IM, Mydlarz WK, Mithani SK, Califano JA (2007) DNA global hypomethylation in squamous cell head and neck cancer associated with smoking, alcohol consumption and stage. Int J Cancer 121:1724–1728
Smith IM, Mithani SK, Mydlarz WK, Chang SS, Califano JA (2010) Inactivation of the tumor suppressor genes causing the hereditary syndromes predisposing to head and neck cancer via promoter hypermethylation in sporadic head and neck cancers. ORL J Otorhinolaryngol Relat Spec 72:44–50
Smollich M, Wülfing P (2008) Targeting the endothelin system: novel therapeutic options in gynecological, urological and breast cancers. Expert Rev Anticancer Ther 8:1481–1493
Sogabe Y, Suzuki H, Toyota M, Ogi K, Imai T, Nojima M, Sasaki Y, Hiratsuka H, Tokino T (2008) Epigenetic inactivation of SFRP genes in oral squamous cell carcinoma. Int J Oncol 32:1253–1261
Steinmann K, Sandner A, Schagdarsurengin U, Dammann RH (2009) Frequent promoter hypermethylation of tumor-related genes in head and neck squamous cell carcinoma. Oncol Rep 22:1519–1526
Subbalekha K, Pimkhaokham A, Pavasant P, Chindavijak S, Phokaew C, Shuangshoti S, Matangkasombut O, Mutirangura A (2009) Detection of LINE-1s hypomethylation in oral rinses of oral squamous cell carcinoma patients. Oral Oncol 45:184–191
Sun D, Zhang Z, Van do N, Huang G, Ernberg I, Hu L (2007) Aberrant methylation of CDH13 gene in nasopharyngeal carcinoma could serve as a potential diagnostic biomarker. Oral Oncol 43:82–87
Sun W, Liu Y, Glazer CA, Shao C, Bhan S, Demokan S, Zhao M, Rudek MA, Ha PK, Califano JA (2010) TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1alpha stabilization. Clin Cancer Res 16:857–866
Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, Pretlow TP, Yang B, Akiyama Y, Van Engeland M, Toyota M, Tokino T, Hinoda Y, Imai K, Herman JG, Baylin SB (2004) Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet 36:417–422
Suzuki E, Imoto I, Pimkhaokham A, Nakagawa T, Kamata N, Kozaki KI, Amagasa T, Inazawa J (2007) PRTFDC1, a possible tumor-suppressor gene, is frequently silenced in oral squamous-cell carcinomas by aberrant promoter hypermethylation. Oncogene 26:7921–7932
Szpakowski S, Sun X, Lage JM, Dyer A, Rubinstein J, Kowalski D, Sasaki C, Costa J, Lizardi PM (2009) Loss of epigenetic silencing in tumors preferentially affects primate-specific retroelements. Gene 448:151–167
Tabor MP, Brakenhoff RH, van Houten VM, Kummer JA, Snel MH, Snijders PJ, Snow GB, Leemans CR, Braakhuis BJ (2001) Persistence of genetically altered fields in head and neck cancer patients: biological and clinical implications. Clin Cancer Res 7:1523–1532
Tabor MP, Brakenhoff RH, Ruijter-Schippers HJ, Van Der Wal JE, Snow GB, Leemans CR, Braakhuis BJ (2002) Multiple head and neck tumors frequently originate from a single preneoplastic lesion. Am J Pathol 161:1051–1060
Tabor MP, Brakenhoff RH, Ruijter-Schippers HJ, Kummer JA, Leemans CR, Braakhuis BJ (2004) Genetically altered fields as origin of locally recurrent head and neck cancer: a retrospective study. Clin Cancer Res 10:3607–3613
Taioli E, Ragin C, Wang XH, Chen J, Langevin SM, Brown AR, Gollin SM, Garte S, Sobol RW (2009) Recurrence in oral and pharyngeal cancer is associated with quantitative MGMT promoter methylation. BMC Cancer 9:354
Takeshima M, Saitoh M, Kusano K, Nagayasu H, Kurashige Y, Malsantha M, Arakawa T, Takuma T, Chiba I, Kaku T, Shibata T, Abiko Y (2008) High frequency of hypermethylation of p14, p15 and p16 in oral pre-cancerous lesions associated with betel-quid chewing in Sri Lanka. J Oral Pathol Med 37:475–479
Tan SH, Ida H, Goh BC, Hsieh W, Loh M, Ito Y (2006) Analyses of promoter hypermethylation for RUNX3 and other tumor suppressor genes in nasopharyngeal carcinoma. Anticancer Res 26:4287–4292
Tan HK, Saulnier P, Auperin A, Lacroix L, Casiraghi O, Janot F, Fouret P, Temam S (2008) Quantitative methylation analyses of resection margins predict local recurrences and disease-specific deaths in patients with head and neck squamous cell carcinomas. Br J Cancer 99:357–363
Taniguchi H, Yamamoto H, Hirata T, Miyamoto N, Oki M, Nosho K, Adachi Y, Endo T, Imai K, Shinomura Y (2005) Frequent epigenetic inactivation of Wnt inhibitory factor-1 in human gastrointestinal cancers. Oncogene 24:7946–7952
Tao J, Wu B, Fang W (1997) Mutation and methylation of CDKN2 gene in human head and neck carcinomas. Zhonghua Bing Li Xue Za Zhi 26:152–154
Tokumaru Y, Yamashita K, Osada M, Nomoto S, Sun DI, Xiao Y, Hoque MO, Westra WH, Califano JA, Sidransky D (2004) Inverse correlation between cyclin A1 hypermethylation and p53 mutation in head and neck cancer identified by reversal of epigenetic silencing. Cancer Res 64:5982–5987
Tokumaru Y, Yahata Y, Fujii M (2005) Aberrant promoter hypermethylation of tazarotine-induced gene 1 (TIG1) in head and neck cancer. Nippon Jibiinkoka Gakkai Kaiho 108:1152–1157
Tokumaru Y, Yamashita K, Kim MS, Park HL, Osada M, Mori M, Sidransky D (2008) The role of PGP9.5 as a tumor suppressor gene in human cancer. Int J Cancer 123:753–759
Tong JH, Ng DC, Chau SL, So KK, Leung PP, Lee TL, Lung RW, Chan MW, Chan AW, Lo KW, To KF (2010) Putative tumour-suppressor gene DAB2 is frequently downregulated by promoter hypermethylation in nasopharyngeal carcinoma. BMC Cancer 10:253
Toyota M, Sasaki Y, Satoh A, Ogi K, Kikuchi T, Suzuki H, Mita H, Tanaka N, Itoh F, Issa JP, Jair KW, Schuebel KE, Imai K, Tokino T (2003) Epigenetic inactivation of CHFR in human tumors. Proc Natl Acad Sci USA 100:7818–7823
Tsao SW, Liu Y, Wang X, Yuen PW, Leung SY, Yuen ST, Pan J, Nicholls JM, Cheung AL, Wong YC (2003) The association of E-cadherin expression and the methylation status of the E-cadherin gene in nasopharyngeal carcinoma cells. Eur J Cancer 39:524–531
Tsunematsu T, Kudo Y, Iizuka S, Ogawa I, Fujita T, Kurihara H, Abiko Y, Takata T (2009) RUNX3 has an oncogenic role in head and neck cancer. PLoS One 4:e5892
Uchida D, Begum NM, Almofti A, Kawamata H, Yoshida H, Sato M (2004) Frequent downregulation of 14-3-3 sigma protein and hypermethylation of 14-3-3 sigma gene in salivary gland adenoid cystic carcinoma. Br J Cancer 91:1131–1138
Ushijima T (2007) Epigenetic field for cancerization. J Biochem Mol Biol 40:142–150
Viswanathan M, Tsuchida N, Shanmugam G (2003) Promoter hypermethylation profile of tumor-associated genes p16, p15, hMLH1, MGMT and E-cadherin in oral squamous cell carcinoma. Int J Cancer 105:41–46
Wagner JM, Hackanson B, Lübbert M, Jung M (2010) Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics 1:117–136
Wang T, Liu H, Chen Y, Liu W, Yu J, Wu G (2009) Methylation associated inactivation of RASSF1A and its synergistic effect with activated K-Ras in nasopharyngeal carcinoma. J Exp Clin Cancer Res 28:160
Weber A, Bellmann U, Bootz F, Wittekind C, Tannapfel A (2002) INK4a-ARF alterations and p53 mutations in primary and consecutive squamous cell carcinoma of the head and neck. Virchows Arch 441:133–142
Weber A, Hengge UR, Bardenheuer W, Tischoff I, Sommerer F, Markwarth A, Dietz A, Wittekind C, Tannapfel A (2005) SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene 24:6699–6708
Williams MD, Chakravarti N, Kies MS, Maruya S, Myers JN, Haviland JC, Weber RS, Lotan R, El-Naggar AK (2006) Implications of methylation patterns of cancer genes in salivary gland tumors. Clin Cancer Res 12:7353–7358
Wong TS, Chang HW, Tang KC, Wei WI, Kwong DL, Sham JS, Yuen AP, Kwong YL (2002) High frequency of promoter hypermethylation of the death-associated protein-kinase gene in nasopharyngeal carcinoma and its detection in the peripheral blood of patients. Clin Cancer Res 8:433–437
Wong TS, Kwong DL, Sham JS, Tsao SW, Wei WI, Kwong YL, Yuen AP (2003a) Promoter hypermethylation of high-in-normal 1 gene in primary nasopharyngeal carcinoma. Clin Cancer Res 9:3042–3046
Wong TS, Tang KC, Kwong DL, Sham JS, Wei WI, Kwong YL, Yuen AP (2003b) Differential gene methylation in undifferentiated nasopharyngeal carcinoma. Int J Oncol 22:869–874
Worsham MJ, Chen KM, Stephen JK, Havard S, Benninger MS (2010) Novel approaches to global mining of aberrantly methylated promoter sites in squamous head and neck cancer. Otolaryngol Head Neck Surg 143:116–121, 121.e1–121.e19
Xi S, Dyer KF, Kimak M, Zhang Q, Gooding WE, Chaillet JR, Chai RL, Ferrell RE, Zamboni B, Hunt J, Grandis JR (2006) Decreased STAT1 expression by promoter methylation in squamous cell carcinogenesis. J Natl Cancer Inst 98:181–189
Yakushiji T, Noma H, Shibahara T, Arai K, Yamamoto N, Tanaka C, Uzawa K, Tanzawa H (2001) Analysis of a role for p16/CDKN2 expression and methylation patterns in human oral squamous cell carcinoma. Bull Tokyo Dent Coll 42:159–168
Yalniz Z, Demokan S, Suoglu Y, Ulusan M, Dalay N (2011) Simultaneous methylation profiling of tumor suppressor genes in head and neck cancer. DNA Cell Biol 30:17–24
Yamashita K, Upadhyay S, Osada M, Hoque MO, Xiao Y, Mori M, Sato F, Meltzer SJ, Sidransky D (2002) Pharmacologic unmasking of epigenetically silenced tumor suppressor genes in esophageal squamous cell carcinoma. Cancer Cell 2:485–495
Yan PS, Venkataramu C, Ibrahim A, Liu JC, Shen RZ, Diaz NM, Centeno B, Weber F, Leu YW, Shapiro CL, Eng C, Yeatman TJ, Huang TH (2006) Mapping geographic zones of cancer risk with epigenetic biomarkers in normal breast tissue. Clin Cancer Res 12:6626–6636
Yanatatsaneejit P, Chalermchai T, Kerekhanjanarong V, Shotelersuk K, Supiyaphun P, Mutirangura A, Sriuranpong V (2008) Promoter hypermethylation of CCNA1, RARRES1, and HRASLS3 in nasopharyngeal carcinoma. Oral Oncol 44:400–406
Yeh KT, Shih MC, Lin TH, Chen JC, Chang JY, Kao CF, Lin KL, Chang JG (2002) The correlation between CpG methylation on promoter and protein expression of E-cadherin in oral squamous cell carcinoma. Anticancer Res 22:3971–3975
Yeh KT, Chang JG, Lin TH, Wang YF, Tien N, Chang JY, Chen JC, Shih MC (2003) Epigenetic changes of tumor suppressor genes, P15, P16, VHL and P53 in oral cancer. Oncol Rep 10:659–663
Yi HM, Li H, Peng D, Zhang HJ, Wang L, Zhao M, Yao KT, Ren CP (2006) Genetic and epigenetic alterations of LTF at 3p21.3 in nasopharyngeal carcinoma. Oncol Res 16:261–272
Yi HM, Ren CP, Peng D, Zhou L, Li H, Yao KT (2007) Expression, loss of heterozygosity, and methylation of GNAT1 gene in nasopharyngeal carcinoma. Ai Zheng 26:9–14
Yi B, Tan SX, Tang CE, Huang WG, Cheng AL, Li C, Zhang PF, Li MY, Li JL, Yi H, Peng F, Chen ZC, Xiao ZQ (2009) Inactivation of 14-3-3 sigma by promoter methylation correlates with metastasis in nasopharyngeal carcinoma. J Cell Biochem 106:858–866
Ying J, Li H, Seng TJ, Langford C, Srivastava G, Tsao SW, Putti T, Murray P, Chan AT, Tao Q (2006) Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene 25:1070–1080
Youssef EM, Lotan D, Issa JP, Wakasa K, Fan YH, Mao L, Hassan K, Feng L, Lee JJ, Lippman SM, Hong WK, Lotan R (2004) Hypermethylation of the retinoic acid receptor-beta(2) gene in head and neck carcinogenesis. Clin Cancer Res 10:1733–1742
Zhang S, Kong WJ (2004) Hypermethylation of the death-associated protein kinase promoter in laryngeal squamous cell cancer. Zhonghua Zhong Liu Za Zhi 26:469–471
Zhang S, Kong WJ, Liu Z (2004) Promoter hypermethylation of DNA repair gene O6-methylguanine DNA methyltransferase in laryngeal squamous cell carcinoma. Zhonghua Er Bi Yan Hou Ke Za Zhi 39:152–156
Zhang S, Guo C, Kong W, Liu Z (2006) Promoter hypermethylation of DNA repair gene MGMT in laryngeal squamous cell carcinoma. J Huazhong Univ Sci Technolog Med Sci 26:101–104
Zhang CY, Mao L, Li L, Tian Z, Zhou XJ, Zhang ZY, Li J (2007a) Promoter methylation as a common mechanism for inactivating E-cadherin in human salivary gland adenoid cystic carcinoma. Cancer 110:87–95
Zhang Z, Sun D, Van do N, Tang A, Hu L, Huang G (2007b) Inactivation of RASSF2A by promoter methylation correlates with lymph node metastasis in nasopharyngeal carcinoma. Int J Cancer 120:32–38
Zhou L, Jiang W, Ren C, Yin Z, Feng X, Liu W, Tao Q, Yao K (2005) Frequent hypermethylation of RASSF1A and TSLC1, and high viral load of Epstein–Barr virus DNA in nasopharyngeal carcinoma and matched tumor-adjacent tissues. Neoplasia 7:809–815
Zhou L, Feng X, Shan W, Zhou W, Liu W, Wang L, Zhu B, Yi H, Yao K, Ren C (2007) Epigenetic and genetic alterations of the EDNRB gene in nasopharyngeal carcinoma. Oncology 72:357–363
Zhou W, Feng X, Li H, Wang L, Zhu B, Liu W, Zhao M, Yao K, Ren C (2009) Inactivation of LARS2, located at the commonly deleted region 3p21.3, by both epigenetic and genetic mechanisms in nasopharyngeal carcinoma. Acta Biochim Biophys Sin (Shanghai) 41:54–62
Zuo C, Ai L, Ratliff P, Suen JY, Hanna E, Brent TP, Fan CY (2004) O6-methylguanine-DNA methyltransferase gene: epigenetic silencing and prognostic value in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 13:967–975
Zuo C, Zhang H, Spencer HJ, Vural E, Suen JY, Schichman SA, Smoller BR, Kokoska MS, Fan CY (2009) Increased microsatellite instability and epigenetic inactivation of the hMLH1 gene in head and neck squamous cell carcinoma. Otolaryngol Head Neck Surg 141:484–490
Conflict of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Demokan, S., Dalay, N. Role of DNA methylation in head and neck cancer. Clin Epigenet 2, 123–150 (2011). https://doi.org/10.1007/s13148-011-0045-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13148-011-0045-3