
Abstract
Background
Glutathione S-transferase P1 (GSTP1) has been reported to function as a tumor suppressor gene in various types of human cancers. Aberrant methylation of tumor-related genes at the promoter regions can inactivate genes, which is important in the carcinogenesis of breast cancer. However, the role of GSTP1 promoter methylation in the occurrence of breast cancer and its relationship with tumor stage and histological grade has not been fully elucidated. Thus, we carried out a meta-analysis to yield a more accurate association.
Methods
A systematically literature search was made on PubMed, EMBASE and Web of Science databases for eligible studies. The odds ratio (OR) and 95 % confidence interval (95 % CI) were calculated by RevMan 5.2 software. Subgroup and sensitivity analyses were conducted to explore the source of heterogeneity.
Results
Eventually, 17 articles involving 19 case–control studies were included in the present meta-analysis. Overall, the pooled results indicated that aberrant GSTP1 promoter methylation was significantly associated with the risk of breast cancer (OR = 7.85, 95 % CI = 5.12–12.01; Caucasians OR = 7.23, 95 % CI = 3.76–13.90 and Asians OR = 11.71, 95 % CI = 5.69–24.07). Furthermore, our results revealed that GSTP1 promoter methylation was more often observed in late-stage breast cancer patients compared with early-stage ones (OR = 1.84, 95 % CI = 1.32–2.58). However, no significant association was identified between GSTP1 promoter methylation and histological grade (OR = 0.74, 95 % CI = 0.43–1.26).
Conclusions
The results indicated that GSTP1 promoter methylation probably plays an important role in breast carcinogenesis, which could serve as an effective biomarker for the diagnosis and monitor of breast cancer.
Similar content being viewed by others


Background
Breast cancer, a heterogeneous disease, is by far the most common malignancy that affects females. It has been reported that an estimated 1.7 million new cases of breast cancer were diagnosed with nearly 522,000 related deaths worldwide in 2012 [1]. Moreover, incidence rates differ between regions with a lifetime risk of 1 in 3 women in Asia and 1 in 8 women in the United States [2]. Despite intensive research, the molecular mechanism of cancer development is still not fully understood. Generally, the interplay between genetic and environmental risk factors has played an important role in the etiology of breast cancer [3]. In recent years, increasing evidence has shown that epigenetic changes of tumor-related genes are involved in the pathogenesis and development of breast cancer, and could be used as indicators of cancer diagnosis and treatment [4–6].
Glutathione-S-transferases (GSTs) are a family of enzymes involved in the detoxification of carcinogenic and cytotoxic substances by catalyzing their conjugation with reduced glutathione [7, 8]. Among the isoenzymes, the pi-class GST (GSTπ) encoded by the GSTP1 gene is implicated in a large variety of detoxification and metabolism reactions, which prevent cells from genome damage and cancer initiation [9, 10]. The GSTP1 gene is a tumor suppressor gene and locus on chromosome 11q13 [11]. Aberrant methylation of the GSTP1 often occurs in different cancer types including those of liver, prostate, and breast cancer [12, 13]. Moreover, the silencing of GSTP1 gene expression induced by promoter methylation has been found to be implicated in the pathogenesis of breast cancer [14].
To date, several studies have investigated the methylation patterns of the GSTP1 in breast cancer patients, yet the data are greatly variable due to differences among studies. Therefore, we conducted a meta-analysis of the published clinical studies to evaluate the effect of GSTP1 promoter methylation on breast cancer patients.
Methods
Eligible criteria
Eligible studies included in this meta-analysis should meet the following standards: (1) assessed the association between GSTP1 promoter methylation and breast cancer; (2) independent case–control studies; (3) all patients met the clear diagnostic criteria for breast cancer; (4) provided sufficient data about the methylation levels of GSTP1 in tissue or blood samples of cancer patients and normal controls; (5) the methylated GSTP1 was detected by polymerase chain reaction (PCR) based methylation assays.
Literature search
This meta-analysis was reported according to the checklist of the Meta-analysis of Observational Studies in Epidemiology (MOOSE) guidelines (Additional file 1: Table S1). We systematically searched related clinical studies regarding the association between GSTP1 promoter methylation and breast cancer via PubMed, EMBASE and Web of Science databases (up to January 31, 2015). The key terms: (breast OR mammary) And (cancer OR neoplasm OR tumor OR carcinoma) And (GSTP1 OR glutathione S-transferase P1) And (methylation OR hypermethylation) were used. The references cited in the selected studies were also scanned for relevant studies.
Data extraction
Data extraction was conducted independently by two reviewers from the included studies. The recorded information for each study contained the following: First author’s name, year of publication, patients’ ethnicity, sample type, sample size, tumor stage, histological grade, GSTP1 methylation frequencies and the methylation detection methods. All selected studies used normal samples as controls, which were composed of normal breast tissues from breast cancer patients and normal samples from non-cancer people. Of these studies, we combined stage 0, I and II as early-stage, stage III and IV as late-stage, which were defined by AJCC staging system [15]. As for histological grade, Grade I and II were defined as low-grade, Grade III was defined as high-grade [16].
Statistical analysis
Odds ratios (ORs) and their 95 % confidence intervals (CIs) were used to evaluate the association. Heterogeneity was quantified by the Cochran Q test with statistical significance set at P < 0.10 and I2 statistics. If there was no statistical heterogeneity among studies (P ≥ 0.10 and I2 < 40 %), we used the fixed-effect model to pool the results; otherwise, the random-effects model was applied [17]. Moreover, subgroup analyses, which were stratified according to the patients’ ethnicity, sample type and detection methods were performed to explore potential sources of heterogeneity and the differences among them. In the presence of heterogeneity, sensitivity analysis was conducted by omitting a single study in each turn to see whether a particular omission could influence the overall estimate. The funnel plots were applied to assess publication bias if the included number of studies was no less than nine. All above analyses were carried out using the Review Manager 5.2 software (Cochrane Collaboration, Oxford, UK). In addition, the effect of possible publication bias was evaluated using the Egger’s test [18] and trim-and-fill method [19] by STATA 12.0 software.
Results
Studies selection and characteristics
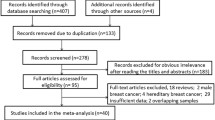
After being selected in accordance with our inclusion criteria, 17 articles involving 19 case–control studies [20–36], comprising 1,647 cases and 559 controls were finally included, the publication years of the selected studies ranged from 2003–2014. Figure 1 showed the process of study selection.

The flow diagram of the study selection process
Ten studies were conducted among Caucasians [20, 21, 24–26, 28, 30, 32, 35, 36], seven studies among Asians [23, 29, 31, 33, 34], one study among Africa [22] and the other one was among mixed populations [27]. Tumor tissues and blood samples were used to detect the methylation status of GSTP1 promoter. Moreover, the methylated levels of GSTP1 were assessed using a variety of PCR based methylation assays composing of methylation-specific PCR (MSP), quantitative MSP (QMSP), pyrosequencing, MethyLight assay, and methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA). Characteristics of all included studies were summarized in Table 1.
Overall and subgroup analyses
Our results showed that breast cancer exhibited significantly higher frequency of GSTP1 methylation than normal controls (OR = 7.85, 95 % CI = 5.12–12.01, Fig. 2). Moreover, subgroup analyses were performed to identify the influence of abnormal GSTP1 promoter methylation on the risk of breast cancer. Ethnicity-stratified analysis revealed that there were statistical associations between GSTP1 promoter methylation and increased breast cancer risk among both Caucasians (OR = 7.23, 95 % CI = 3.76–13.90) and Asians (OR = 11.71, 95 % CI = 5.69–24.07). After stratified by sample type, we found that aberrant methylation of GSTP1 was correlated with the risk of breast cancer detected in tissue (OR = 10.32, 95 % CI = 5.97–17.85) as well as blood samples (OR = 4.02, 95 % CI = 1.12–14.38). After stratified by method, significant associations between GSTP1 promoter methylation and the risk of breast cancer were observed in all of the subgroups (Quantitative: OR = 4.73, 95 % CI = 1.84–12.12; Semi-quantitative: OR = 10.33, 95 % CI = 3.32–32.10; Non-quantitative: OR = 12.55, 95 % CI = 5.72–27.55). Above results could be reviewed in Table 2.

Forest plot of the association between GSTP1 methylation and breast cancer risk based on a fixed-effect model. The squares and horizontal lines correspond to the OR and 95 % CI
In addition, eight studies [22, 28, 29, 33–35] comprising 832 patients were pooled for the OR in evaluating the association between GSTP1 methylation and tumor stage. The results revealed that aberrant GSTP1 methylation was more often observed in late-stage patients compared with early-stage ones (OR = 1.84, 95 % CI = 1.32–2.58, Fig. 3). However, the pooled OR of five studies showed that there was no significant association between GSTP1 methylation and histological grade (OR = 0.74, 95 % CI = 0.43–1.26) (Table 3).

Forest plot of the association between GSTP1 methylation and tumor stage based on a fixed-effect model. OR and 95 % CI were calculated
Sensitivity analysis
The results showed that moderate heterogeneity existed in investigating the correlation of GSTP1 methylation and breast cancer risk detected in blood samples and quantitative method by subgroup analysis (Table 2). Then, a sensitive analysis was used to find the heterogeneous study. After removal of the study by Brooks et al. [27], the heterogeneity presented in blood samples was reduced from I2 = 54 % (Ph = 0.07) to I2 = 0 % (Ph = 0.71), the heterogeneity in quantitative method was also reduced from I2 = 45 % (Ph = 0.08) to I2 = 0 % (Ph = 0.43), suggesting it might be the heterogeneous study. However, the pooled ORs were not significantly changed in sensitivity analyses, in which each study was deleted at one time, suggested the stability of our results.
Publication bias
Visual inspection of the funnel plot in Fig. 4 shows an asymmetry, which indicates the presence of publication bias in evaluating GSTP1 methylation and breast cancer risk. Egger’s test also display statistical evidence of asymmetry (P = 0.003). Then, the trim-and-fill method was applied to adjust this bias and calculate the number of unpublished studies that could lead to asymmetry (Fig. 5). The estimated OR adjusted by trim-and-fill method was similar to the original estimate (OR = 4.20, 95 % CI = 2.75–6.41), indicating that our analyses were reliable and robust. For limited number of studies, the investigation of publication bias with tumor stage and histological grade were not examined.

Funnel plot for evaluating publication bias test for GSTP1 methylation and breast cancer risk. The standard error of log (OR) of each study was plotted against its log (OR)

Funnel plot of publication bias test for GSTP1 methylation and breast cancer risk after trim-and-fill method. Logor natural logarithm of OR, horizontal line mean effect size
Discussion
GSTP1 is a member of the metabolic enzymes family, which has significant implications in the prevention of cancer initiation upon exposure to carcinogens [11, 13]. Absence of GSTP1 expression is found in approximately two thirds of the patients with breast cancer, suggesting it might play an important role in breast carcinogenesis [23]. It has also been demonstrated that the hypermethylation of GSTP1 gene promoter frequently occurs in breast cancer and may result in inactivation of GSTP1 expression, thus lead to cancer progression [13].
The current meta-analysis demonstrated that the methylation level of GSTP1 was significantly higher in breast cancer patients than that in normal controls, which indicated its potential role in the etiology of breast cancer. This was in accordance with the results of previous studies [22–24]. We also performed subgroup analyses to further explore the potential effects of the patients’ ethnicity, sample type and detection method on the association of GSTP1 promoter methylation with the risk of breast cancer. The results revealed that GSTP1 promoter methylation was closely associated with the risk of breast cancer in both Caucasians (OR = 7.23, 95 % CI = 3.76–13.90) and Asians (OR = 11.71, 95 % CI = 5.69–24.07), whereas, the correlation was stronger in Asians than in Caucasians. The reasons might include differences in genetic backgrounds, environments and sample size. After stratified by sample type, we found that aberrant methylation of GSTP1 was correlated with the risk of breast cancer detected in tissue (OR = 10.32, 95 % CI = 5.97–17.85) as well as blood samples (OR = 4.02, 95 % CI = 1.12–14.38). Moreover, a high concordance between tumor and blood DNA methylation of GSTP1 was reported in studies conducted on paired tumor tissue and blood samples from breast cancer patients [29, 33]. Yamamoto et al. compared the gene methylation status in serum DNA before and after surgery in patients with primary breast cancer, and demonstrated that the origin of blood methylated DNA was the tumor tissue because patients with aberrant GSTP1 methylation in serum DNA collected before surgery were found to be negative for gene methylation after surgery [33]. This indicated that blood DNA methylation of GSTP1 could reflect alterations in the tumor and the ease of obtaining blood samples makes it a potential biomarker for diagnosis of breast cancer. In the present meta-analysis, the small number of patients, various ethnicity groups and different time of sample collection may contribute to relatively extended confidence intervals. To date, a diversity of PCR based methylation assays were developed and widely used to measure methylation in clinical specimens, classified as quantitative, semi-quantitative and non-quantitative techniques [37]. Several papers have compared MS–MLPA (semi-quantitative) with pyrosequencing (quantitative) or MSP (non-quantitative) and showed a good concordance between MS–MLPA and pyrosequencing [30, 37]. Since different methylation assays were applied to detect the methylation levels of GSTP1 in the studies included in this meta-analysis, we also performed subgroup analysis based on methods to explore potential sources of heterogeneity and the differences among them. As a result, significant associations were identified as detected by quantitative, semi-quantitative and non-quantitative techniques, suggested these methods have the same effect in GSTP1 methylation detection. However, when only quantitative analyses of GSTP1 promoter methylation in blood DNA are pooled, no significant association was observed (data not shown). It is hypothesized that the small sample size may lead to false-negative results. Furthermore, different patient materials and the choice of different primer sets between different studies may influence the results. Aberrantly methylated genes are frequently found in human cancers but rarely in normal controls, and their presence is not an exclusive attribute of metastatic cancer. Examination of body fluid from patients with early stage or organ-confined tumors may also reveal positive results [28]. Our study showed that the methylation level of GSTP1 increased significantly in late-stage compared to the early stage breast carcinomas, suggested that breast cancer patients with GSTP1 promoter hypermethylation may have a biologically aggressive phenotype.
Breast cancer is a complex multifactorial disease that is driven by genetic and epigenetic alterations, which cause aberrant gene function [38]. Previous study declared that the genetic variation of GSTP1 affected its enzymatic activity and detoxifcation ability, thereby contributing to breast cancer susceptibility [10]. Epigenetic alterations including DNA methylation and histone modifications which occur in transformed cells are identified as an early event during tumor development [35, 39]. In breast cancer, hypermethylation of promoter CpG islands has been described as the main epigenetic pathway to inactivate genes involved in various aspects of cellular function [30]. It has been reported that GSTP1 is capable of inhibiting tumor growth by its interaction with the c-Jun N-terminal kinase (JNK1) signaling [11], suggesting its role as a tumor suppressor gene. Additionally, because of its detoxifying effects on the anticancer agents, GSTP1 may also affect the sensitivity of breast tumors to chemotherapy, emerged as a novel therapeutic target [7, 9].
To our knowledge, this is the first meta-analysis comprehensively performed to assess the relationship between GSTP1 promoter methylation and the incidence of breast cancer. Nevertheless, a number of potential limitations should be acknowledged. First, the effects of potential risk factors such as age, subtype and hormone receptor status on the current results of this meta-analysis could not be eliminated for lack of detailed information. Second, meta-analysis is a secondary analysis and the heterogeneity is the major issue in genetic studies [40–43]. Of course, our meta-analysis also suffered this issue and we performed subgroup analyses to explore the origin. The results showed that different methylation methods, source of controls and cut-offs positivity of hypermethylation might contribute to heterogeneity. Third, only published clinical studies were selected in this meta-analysis, some unpublished and negative studies may contribute to publication bias. Since studies with statistically positive results were easier to publish than those with negative results, publication bias is inevitable. However, the estimated OR adjusted for publication bias by trim-and-fill method was not substantially changed. Fourth, although our initial search has no language restrictions, only articles published in English and Chinese finally were reviewed. This due to the language ability and the right to use databases of our team, and also might result in some bias.
Conclusions
In conclusion, our meta-analysis suggested a strong association between GSTP1 promoter methylation and breast cancer risk. Thus, aberrant GSTP1 promoter methylation could be a helpful biomarker for the early screening of breast cancer. However, given the limitations elaborated above, high quality studies with larger sample sizes should be employed in further research.
Abbreviations
- AJCC:
-
American Joint Committee on Cancer
- CI:
-
Confidence interval
- GSTP1:
-
glutathione s-transferase P1
- MOOSE:
-
Meta-analysis of Observational Studies in Epidemiology
- MS-MLPA:
-
methylation-specific multiplex ligation-dependent probe amplification
- MSP:
-
methylation-specific polymerase chain reaction
- OR:
-
odds ratio
- QMSP:
-
quantitative methylation-specific polymerase chain reaction
- RevMan:
-
Review Manager
References
Tao Z, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast Cancer: Epidemiology and Etiology. Cell Biochem Biophys. 2015;72(2):333-8.
Donepudi MS, Kondapalli K, Amos SJ, Venkanteshan P. Breast cancer statistics and markers. J Cancer Res Ther. 2014;10(3):506–11.
Widschwendter M, Jones PA. DNA methylation and breast carcinogenesis. Oncogene. 2002;21(35):5462–82.
Barekati Z, Radpour R, Lu Q, Bitzer J, Zheng H, Toniolo P, et al. Methylation signature of lymph node metastases in breast cancer patients. BMC Cancer. 2012;12:244.
Radpour R, Barekati Z, Kohler C, Lv Q, Burki N, Diesch C, et al. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS One. 2011;6(1):e16080.
Chen KM, Stephen JK, Raju U, Worsham MJ. Delineating an epigenetic continuum for initiation, transformation and progression to breast cancer. Cancers (Basel). 2011;3(2):1580–92.
Miyake T, Nakayama T, Naoi Y, Yamamoto N, Otani Y, Kim SJ, et al. GSTP1 expression predicts poor pathological complete response to neoadjuvant chemotherapy in ER-negative breast cancer. Cancer Sci. 2012;103(5):913–20.
Jhaveri MS, Morrow CS. Methylation-mediated regulation of the glutathione S-transferase P1 gene in human breast cancer cells. Gene. 1998;210(1):1–7.
Esteller M, Corn PG, Urena JM, Gabrielson E, Baylin SB, Herman JG. Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res. 1998;58(20):4515–8.
Saxena A, Dhillon VS, Shahid M, Khalil HS, Rani M, Prasad DT, et al. GSTP1 methylation and polymorphism increase the risk of breast cancer and the effects of diet and lifestyle in breast cancer patients. Exp Ther Med. 2012;4(6):1097–103.
Arai T, Miyoshi Y, Kim SJ, Taguchi T, Tamaki Y, Noguchi S. Association of GSTP1 CpG islands hypermethylation with poor prognosis in human breast cancers. Breast Cancer Res Treat. 2006;100(2):169–76.
Krassenstein R, Sauter E, Dulaimi E, Battagli C, Ehya H, Klein-Szanto A, et al. Detection of breast cancer in nipple aspirate fluid by CpG island hypermethylation. Clin Cancer Res. 2004;10(1 Pt 1):28–32.
Pongtheerat T, Pakdeethai S, Purisa W, Chariyalertsak S, Petmitr S. Promoter methylation and genetic polymorphism of glutathione S-transferase P1 gene (GSTP1) in Thai breast- cancer patients. Asian Pac J Cancer Prev. 2011;12(10):2731–4.
Otani Y, Miyake T, Kagara N, Shimoda M, Naoi Y, Maruyama N, et al. BRCA1 promoter methylation of normal breast epithelial cells as a possible precursor for BRCA1-methylated breast cancer. Cancer Sci. 2014;105(10):1369–76.
Singletary SE, Allred C, Ashley P, Bassett LW, Berry D, Bland KI, et al. Staging system for breast cancer: revisions for the 6th edition of the AJCC Cancer Staging Manual. Surg Clin North Am. 2003;83(4):803–19.
Elston CW, Ellis IO. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. 2002;41(3A):154–61.
Li S, Zeng XT, Ruan XL, Weng H, Liu TZ, Wang X, et al. Holmium laser enucleation versus transurethral resection in patients with benign prostate hyperplasia: an updated systematic review with meta-analysis and trial sequential analysis. PLoS One. 2014;9(7):e101615.
Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629–34.
Duval S, Tweedie R. Trim and fill: A simple funnel-plot-based method of testing and adjusting for publication bias in meta-analysis. Biometrics. 2000;56(2):455–63.
Jeronimo C, Costa I, Martins MC, Monteiro P, Lisboa S, Palmeira C, et al. Detection of gene promoter hypermethylation in fine needle washings from breast lesions. Clin Cancer Res. 2003;9(9):3413–7.
Shinozaki M, Hoon DS, Giuliano AE, Hansen NM, Wang HJ, Turner R, et al. Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin Cancer Res. 2005;11(6):2156–62.
Hoque MO, Feng Q, Toure P, Dem A, Critchlow CW, Hawes SE, et al. Detection of aberrant methylation of four genes in plasma DNA for the detection of breast cancer. J Clin Oncol. 2006;24(26):4262–9.
Lee JS. GSTP1 promoter hypermethylation is an early event in breast carcinogenesis. Virchows Arch. 2007;450(6):637–42.
Pasquali L, Bedeir A, Ringquist S, Styche A, Bhargava R, Trucco G. Quantification of CpG island methylation in progressive breast lesions from normal to invasive carcinoma. Cancer Lett. 2007;257(1):136–44.
Jeronimo C, Monteiro P, Henrique R, Dinis-Ribeiro M, Costa I, Costa VL, et al. Quantitative hypermethylation of a small panel of genes augments the diagnostic accuracy in fine-needle aspirate washings of breast lesions. Breast Cancer Res Treat. 2008;109(1):27–34.
Hoque MO, Prencipe M, Poeta ML, Barbano R, Valori VM, Copetti M, et al. Changes in CpG islands promoter methylation patterns during ductal breast carcinoma progression. Cancer Epidemiol Biomarkers Prev. 2009;18(10):2694–700.
Brooks JD, Cairns P, Shore RE, Klein CB, Wirgin I, Afanasyeva Y, et al. DNA methylation in pre-diagnostic serum samples of breast cancer cases: results of a nested case–control study. Cancer Epidemiol. 2010;34(6):717–23.
Matuschek C, Bolke E, Lammering G, Gerber PA, Peiper M, Budach W, et al. Methylated APC and GSTP1 genes in serum DNA correlate with the presence of circulating blood tumor cells and are associated with a more aggressive and advanced breast cancer disease. Eur J Med Res. 2010;15:277–86.
Sharma G, Mirza S, Parshad R, Srivastava A, Gupta SD, Pandya P, et al. Clinical significance of promoter hypermethylation of DNA repair genes in tumor and serum DNA in invasive ductal breast carcinoma patients. Life Sci. 2010;87(3–4):83–91.
Moelans CB, Verschuur-Maes AH, van Diest PJ. Frequent promoter hypermethylation of BRCA2, CDH13, MSH6, PAX5, PAX6 and WT1 in ductal carcinoma in situ and invasive breast cancer. J Pathol. 2011;225(2):222–31.
Park SY, Kwon HJ, Lee HE, Ryu HS, Kim SW, Kim JH, et al. Promoter CpG island hypermethylation during breast cancer progression. Virchows Arch. 2011;458(1):73–84.
Kornegoor R, Moelans CB, Verschuur-Maes AH, Hogenes M, de Bruin PC, Oudejans JJ, et al. Promoter hypermethylation in male breast cancer: analysis by multiplex ligation-dependent probe amplification. Breast Cancer Res. 2012;14(4):R101.
Yamamoto N, Nakayama T, Kajita M, Miyake T, Iwamoto T, Kim SJ, et al. Detection of aberrant promoter methylation of GSTP1, RASSF1A, and RARbeta2 in serum DNA of patients with breast cancer by a newly established one-step methylation-specific PCR assay. Breast Cancer Res Treat. 2012;132(1):165–73.
Jung EJ, Kim IS, Lee EY, Kang JE, Lee SM, Kim DC, et al. Comparison of methylation profiling in cancerous and their corresponding normal tissues from korean patients with breast cancer. Ann Lab Med. 2013;33(6):431–40.
Klajic J, Fleischer T, Dejeux E, Edvardsen H, Warnberg F, Bukholm I, et al. Quantitative DNA methylation analyses reveal stage dependent DNA methylation and association to clinico-pathological factors in breast tumors. BMC Cancer. 2013;13:456.
de Groot JS, Pan X, Meeldijk J, van der Wall E, van Diest PJ, Moelans CB. Validation of DNA promoter hypermethylation biomarkers in breast cancer--a short report. Cell Oncol (Dordr). 2014;37(4):297–303.
Suijkerbuijk KP, Pan X, van der Wall E, van Diest PJ, Vooijs M. Comparison of different promoter methylation assays in breast cancer. Anal Cell Pathol (Amst). 2010;33(3):133–41.
Lo PK, Sukumar S. Epigenomics and breast cancer. Pharmacogenomics. 2008;9(12):1879–902.
Tserga A, Michalopoulos NV, Levidou G, Korkolopoulou P, Zografos G, Patsouris E, et al. Association of aberrant DNA methylation with clinicopathological features in breast cancer. Oncol Rep. 2012;27(5):1630–8.
Yan Y, Weng H, Shen ZH, Wu L, Zeng XT. Association between interleukin-4 gene −590 c/t, −33 c/t, and 70-base-pair polymorphisms and periodontitis susceptibility: a meta-analysis. J Periodontol. 2014;85(11):e354–362.
Zeng X, Zhang Y, Kwong JS, Zhang C, Li S, Sun F, et al. The methodological quality assessment tools for preclinical and clinical studies, systematic review and meta-analysis, and clinical practice guideline: a systematic review. J Evid Based Med. 2015;8(1):2–10.
Zeng XT, Leng WD, Zhang C, Liu J, Cao SY, Huang W. Meta-analysis on the association between toothbrushing and head and neck cancer. Oral Oncol. 2015;51(5):446–51.
Zeng XT, Liu DY, Kwong JS, Leng WD, Xia LY, Mao M. Meta-Analysis of Association Between Interleukin-1beta C-511T Polymorphism and Chronic Periodontitis Susceptibility. J Periodontol. 2015;86(6):812-9.
Acknowledgements
The present study was sponsored by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, and was supported by grants from the Natural Science Foundation of China (nos. 30873044 and 81272372).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no conflicts of interest in this work.
Authors’ contributions
FC and LXH conceived the study. FC and WH searched the databases and extracted the data. WXM and WFB assembled and analyzed the data.WXM and ZXT gave advice on meta-analysis methodology. FC wrote the draft of the paper. WXM, ZXT and LXH reviewed the manuscript. All authors have read and approved the final manuscript.
Cheng Fang and Xue-Mei Wei contributed equally to this work.
Additional file
Additional file 1: Table S1.
MOOSE checklist in current meta-analysis. (DOC 73 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fang, C., Wei, XM., Zeng, XT. et al. Aberrant GSTP1 promoter methylation is associated with increased risk and advanced stage of breast cancer: a meta-analysis of 19 case-control studies. BMC Cancer 15, 920 (2015). https://doi.org/10.1186/s12885-015-1926-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-015-1926-1