
Abstract
Background
Invasion and metastasis are two important hallmarks of malignant tumors caused by complex genetic and epigenetic alterations. The present study investigated the contribution of aberrant methylation profiles of cancer related genes, APC, BIN1, BMP6, BRCA1, CST6, ESR-b, GSTP1, P14 (ARF), P16 (CDKN2A), P21 (CDKN1A), PTEN, and TIMP3, in the matched axillary lymph node metastasis in comparison to the primary tumor tissue and the adjacent normal tissue from the same breast cancer patients to identify the potential of candidate genes methylation as metastatic markers.
Methods
The quantitative methylation analysis was performed using the SEQUENOM’s EpiTYPER™ assay which relies on matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS).
Results
The quantitative DNA methylation analysis of the candidate genes showed higher methylation proportion in the primary tumor tissue than that of the matched normal tissue and the differences were significant for the APC, BIN1, BMP6, BRCA1, CST6, ESR-b, P16, PTEN and TIMP3 promoter regions (P<0.05). Among those candidate methylated genes, APC, BMP6, BRCA1 and P16 displayed higher methylation proportion in the matched lymph node metastasis than that found in the normal tissue (P<0.05). The pathway analysis revealed that BMP6, BRCA1 and P16 have a role in prevention of neoplasm metastasis.
Conclusions
The results of the present study showed methylation heterogeneity between primary tumors and metastatic lesion. The contribution of aberrant methylation alterations of BMP6, BRCA1 and P16 genes in lymph node metastasis might provide a further clue to establish useful biomarkers for screening metastasis.
Similar content being viewed by others


Background
Breast cancer is one of the most common malignancies with a high mortality rate among women [1]. Breast cancer, a heterogeneous disease, presents various pathological signs such as axillary lymph node metastasis which is associated with a high risk of recurrence and considered as an important prognosis factor in the early stages of the disease [2, 3]. Invasion and metastasis are two important hallmarks of malignant tumors associated with complex genetic and epigenetic alterations that allow tumors to disseminate throughout lymphatics or blood vessels, giving rise to the colonization and growth of metastatic cells in distant organs [4–6]. Considering that tumor dissemination is an early event in breast cancer [7], genetic and epigenetic analysis of tumors and metastatic lesions could provide results for biomarker discovery and may improve diagnosis, prognosis and proper management of the treatment for breast cancer patients.
The contribution of aberrant DNA hypermethylation of cancer related genes to the transcriptional silencing and carcinogenesis has been demonstrated in different diseases including different cancer types [7, 8]. The methylation profile of genes involved in critical molecular processes such as cell cycle control, DNA repair and angiogenesis in breast cancer has been investigated [9–12]. Since the lymphatic system has a direct dispatch to spread primary tumor cells to the lymph nodes in breast cancer, primary tumor signatures has been considered as surrogate for lymph node metastasis. However, this persuasion has recently been controversial especially in the context of DNA methylation pattern from primary to metastasis in breast cancer [9, 10, 13]. According to best of our knowledge genome-wide DNA methylation was reported in few metastatic breast cancer cell lines ,however, methylation pattern of individual candidate genes in both the primary and lymph node metastases has been explored in clinical specimens [14, 15]. Therefore the profile of metastasis in breast cancer patients is less characterized and poorly understood which needs further studies to understand the relationship between epigenetic alteration and metastases in breast cancer.
Our previous approach revealed methylation signatures of 42, 528 CpG sites from 22 breast cancer candidate genes that demonstrated promoter hypermethylation of 10 genes involved in cell cycle and DNA repair, invasion and metastasis, cell proliferation, signal transduction and cell detoxification [16]. Moreover, we have shown hypermethylation of the two additional genes involved in TP53 regulatory pathway in the breast cancer patients [17, 18]. The significance of using these hypermethylated genes as circulating biomarkers has been explored as well [18]. The present study investigated the contribution of significant aberrant methylation profile of twelve cancer related genes from the aforementioned studies (APC, BIN, BMP6, BRCA1, CST6, ESR-b, GSTP1, P14, P16, P21, PTEN and TIMP3) in matched axillary lymph node metastasis in comparison to the primary tumor tissue and the adjacent normal tissue from the same breast cancer patients to identify the potential of aberrant methylation profile of the candidate genes as metastatic signature.
Methods
Sampling and pathological classification
The study was approved by the local institutional review board (Ethic commission beider Basel, Sichuan University China). Written consent forms were collected from all patients who were involved in this study. Staging and grading was evaluated according to the WHO histological classification. DNA was isolated from 65 samples including matched primary tumors tissue, matched normal tissue and their matched lymph node metastasis of 24 chines patients with breast cancer. The present cohort included 17 matched normal breast tissues that were collected at least 4 cm away from the tumor site and were confirmed as normal tissue by pathologist. The axillary lymph nodes were removed at the same surgery. Part of the samples was embedded using OCT (Optimal Cutting Temperature, Sakura Finetek, U.S.A) and stored in liquid nitrogen. The above procedures were completed within 20 minutes after peeling preparation. The samples were then stored at -80o. The frozen tissues were sectioned in 4 μm thickness and were submitted for hematotoxylin and eosin staining study. According to pathological tumor type and immunohistochemistry staining, studied cohort consisted of patient’s with Invasive Ductal Carcinoma (IDC) and Invasive Lobular Carcinoma (ILC). Breast cancer characteristics, such as staging, histological grading, and hormone receptor expression from the breast cancer patients are listed in Table 1.
The entirely neoplastic and adjacent normal frozen sections were subjected for DNA extraction. Five to ten sections with 90% neoplastic coverage applied for DNA extraction, and last section was checked for the correct characterization, using the High Pure PCR Template Preparation Kit (Roche, Germany). The rest of the samples were fixed in 10% buffered formalin for immunohistochemical staining study.
The analysis for the detection of estrogen receptor (ER), progesterone receptor (PR) and HER2/neu (C-ErbB-2) proteins was carried out using primary antibodies for Rabbit anti-human estrogen receptor monoclonal antibody (clone sp1, Roche), Rabbit anti-human progesterone receptor monoclonal antibody (clone sp2, Roche), Rabbit anti-human HER-2/neu monoclonal antibody (clone 4B5, Roche). Then slide stainings were processed using ultraView Universal DAB Detection Kit (Ventana Medical Systems Inc, Tucson, AZ) (Figure 1). The ER, PR and HER-2/neu were analyzed following ASCO/CAP Guidelines.

Hematotoxylin and eosin staining (H&E) and immunohistochemical staining for estrogen receptor (ER), progesterone receptor (PR) and HER2/neu proteins (400X). (a) Ductal carcinoma. (b) Lobular carcinoma.
Methylation analysis using thymidine-specific cleavage mass array on MALDI-TOF silico-chip
The SEQUENOM’s EpiTYPER™ assay is a methylation quantification method which relies on MALDI-TOF MS [19]. The theory and practice of this approach for DNA methylation quantification has been confirmed by previous studies [16, 20, 21]. In the current study, this assay was used to quantify the methylation of APC, BIN1, BRCA1, BMP6, CST6, ESR-b, GSTP1, P14, P16, P21, PTEN and TIMP3 promoter regions.
Bisulfite treatment
Bisulfite conversion of the target sequences was performed according to the instruction of the Epitect® Bisulfite Kit (QIAGEN AG, Basel, Switzerland).
Primer designing and PCR-tagging for EpiTYPERTM assay
To design PCR primer for the candidate genes, CpG density and CpG sites of the twelve targeted sequences were analyzed. According to our previous publications, we used the same primer sequences which were tagged with T7-promoter for the reverse and a 10 mer sequences to the forward primer to balance the PCR condition and primer pairs can cover the promoter regions with the most CpG sites using MethPrimer. The primer sequences, annealing temperatures (T a ) and PCR conditions are described in Additional file 1.
In vitro transcription, T-cleavage assay and mass spectrometry
Unincorporated dNTPs were dephosphorylated by adding 1.7μL H2O and 0.3 units of shrimp alkaline phosphatase (SAP; SEQUENOM, Inc., San Diego, CA). The reaction mixture was incubated at 37°C for 20 minutes and the SAP was then heat inactivated for 10 minutes at 85°C. Typically, 2μL of the PCR were used directly as a template in a 5μL transcription reaction. Twenty units of T7 R&DNA polymerase (Epicentre, Madison, WI) were used to incorporate dTTP in the transcripts. Ribonucleotides were used at 1 mmol/L and the dNTP substrate at 2.5 mmol/L. In the same step, the in vitro transcription RNase A (SEQUENOM) was added to cleave the in vitro transcript (T-cleavage assay). The mixture was further diluted with H2O to a final volume of 27μL. Twenty-two nanoliters of cleavage reaction were robotically dispensed (nanodispenser) onto silicon chips preloaded with matrix (SpectroCHIP; SEQUENOM, San Diego). Mass spectra data were collected using a MassARRAY Compact MALDI-TOF (SEQUENOM) and spectra’s methylation proportions were generated by the Epityper software v1.0 (SEQUENOM, San Diego).
Cell signaling and pathway analysis
Gene networks and canonical pathways displaying hypermethylated genes in lymph node metastasis were identified using the Pathway Studio® software version 7.1 (Mammal) database (Ariadne Genomics, Inc., Rockville, USA). The functional analysis identified the biological perspective of the genes that were most relevant to the data sets and facilitated the understanding beyond their functional link to breast neoplasm and metastasis.
Statistical methods
Data analysis was performed using the SPSS software (Statistical Software Package for Windows, version 19). Distribution of data was analyzed by Kolmogorov-Smirnov test that demonstrated our data set was not normally distributed (P < 0.001). Quantitative methylation profile of the twelve candidate genes were compared among primary tissue, adjacent matched normal tissue and their matched lymph node metastasis using the two-way hierarchical cluster analysis. The CpG sites for each gene were clustered based on pair-wise Euclidean distances and linkage algorithm for all studied samples according to the previously developed method by Gene Expression Statistical System (GESS) version 7.1.19 (NCSS, Kaysville, Utah, USA) and the statistical differences between mean methylation quantity of the informative CpG sites per genes were identified using Mann-Whitney-U-Test. The non-parametric Spearman's rho test was performed to find out significance clinical-pathological parameters.
Results
Quantitative methylation profiles of the 12 breast cancer candidate genes promoter using MALDI-TOF MS
The methylation profiles of 12 breast cancer candidate genes in matched primary tumors, normal tissue and their matched lymph node metastasis from 24 breast cancer patients were assessed. For all the genes, one amplicon per gene and 250 CpG sites in total per sample (total of 16,250 sites in 65 analyzed samples) were analyzed (Table 2; Additional file 2.)
The different levels of methylation between studied samples were identified using the two-way hierarchical cluster analysis. The methylation cluster analysis for each individual studied gene is illustrated in Additional file 2.
Methylation alterations of the primary tumor tissue
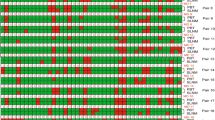
Hierarchical cluster analysis profiling of the promoter alterations and the significance of the alterations are shown in Figure 2(a) for the 12 studied genes.

a) Double dendrogram presents the methylation profiles of the twelve candidate genes in tumor tissue, matched normal tissue and lymph nodes metastasis (Red clusters indicate 0% methylated, yellow clusters indicate 100% methylated, color gradient between red and yellow indicates methylation ranging from 0-100). b) Comparison between quantitative analysis (0%-100%) of methylation for the candidate genes in the studied cohort (* significant correlation; Mann-Whitney U Test).
Methylation quantification of the 12 candidate genes revealed significantly higher methylation proportion for promoter regions of APC, BIN1, BMP6, BRCA1, CST6, ESR-b, P16, PTEN and TIMP3 in primary tumor tissue versus matched normal tissue (P < 0.05, P < 0.05, P < 0.01, P < 0.0001, P < 0.01, P < 0.01, P < 0.05, P < 0.05, P < 0.01 ; respectively). Methylation analysis of GSTP1, P14 and P21 in primary tumor tissue showed slightly higher methylation proportion in comparison to the matched normal tissue, however, the differences were not significant (Figure 2b).
Methylation alterations of the matched lymph node metastasis
Comparison of methylation profiles of the 12 breast cancer candidate genes in the paired lymph node metastasis to the matched normal tissue showed significantly higher methylation levels for APC, BMP6, BRCA1 and P16 genes (P < 0.05 and P < 0.01, P < 0.0001, P < 0.05; respectively). The lymph node metastasis was even more hypermethylated for BMP6 than the primary tumor tissue (P < 0.05). The promoter region of BIN1, BMP6, BRCA1, GSTP1, P14 and TIMP3 showed significantly lower methylation ratio in the lymph node metastasis than the primary tissue ( P < 0.0001, P < 0.05, P < 0.05, P < 0.01, P < 0.05, P < 0.05; respectively).
Comparing the lymph node metastasis to the matched normal tissue, the former revealed differentially lower methylation ratio for BIN1, GSTP1 and P14 promoter regions (P < 0.05, P < 0.01, P < 0.05; respectively) (Figure 2).
Relationship of methylation alterations within the promoter region of 12 breast cancer candidate genes and clinicopathological parameters
Clinicopathological parameters with the methylation proportion of the 12 candidate genes in primary tumor tissue and matched lymph node metastasis were analyzed. The analyses showed significant correlation between higher methylation level of GSTP1 and increasing the histological grade in the primary tumor tissue (Spearman's rho test; P < 0.05). The higher methylation proportion of BIN1 showed significant correlation with expressed ER in primary tumor tissue (Spearman's rho test; P < 0.01). Methylated TIMP3 revealed significant correlation to the primary tumor tissue lacking expression of PR (Spearman's rho test; P < 0.05). The increase of methylation of P21 showed an inverse correlation with the expression of C-ERB2 in the primary tumor tissue (Spearman's rho test; P < 0.05). We could not find any significant correlations between methylation alterations in either of the matched lymph node metastasis and clinicopathological parameters.
Discussion
Aberrant methylation profiles and silencing of a small subset of tumor suppressors and cancer related genes involved in both the primary tumor and lymph node metastasis has been investigated for breast cancer [11, 22–24]. Several studies on breast cancer revealed some similarities and differences between promoter methylation pattern of the studied genes in primary and lymph node metastatic [16, 19, 25]. These investigations highlight the important role of aberrant promoters’ methylation in the metastatic process. In the present study, we implemented the methylation signature of the 12 breast cancer candidate genes (APC, BIN1, BMP6, BRCA1, CST6, ESR-b, GSTP1, P14, P16, P21, PTEN and TIMP3) by comparing lymph nodes metastasis to their matched primary tumor tissues and normal tissues from the same breast cancer patients.
The quantitative methylation analysis of the 12 studied genes in the present cohort showed higher methylation proportion for the primary tumor tissue versus matched normal tissue and the differences were significant for APC, BIN1, BMP6, BRCA1, CST6, ESR-b, P16, PTEN and TIMP3 promoter regions (P < 0.05, P < 0.05, P < 0.01, P < 0.0001, P < 0.01, P < 0.01, P < 0.05, P < 0.05, P < 0.01; respectively). Among the significant methylated genes, APC, BMP6, BRCA1 and P16 represented higher methylation proportions in matched lymph node metastasis compared to those of the normal tissue (P < 0.05 and P < 0.01, P < 0.0001, P < 0.05; respectively). Present findings provided evidence of differences in methylation status between primary tumors and their corresponding matched lymph nodes and demonstrated methylation heterogeneity between primary tumors and metastatic lesion which are in line with previous reports described about primary tumor and metastasis in breast, gastric and colorectal cancers [13, 26, 27]. The results also indicated that some of the cancer specific changes become altered over the metastasis procedure. The mechanism for losing or gaining methylation in lymph nodes metastatic is still not clear while the alteration of the methylation signature from primary to metastasis might be due to the adaptation response of the disseminated cells to the microenvironment at the site of colonization [28].
The molecular function, biological processes and contribution of the studied genes to developing metastasis were analyzed by ResNet® 7 (Mammal). The pathway analysis revealed that BMP6 BRCA1 and P16 have a role in prevention of neoplasm metastasis (Figure 3). The relation of DNA methylation for BRCA1 and P16 with tumor recurrence has been reported with a high value in breast cancer patients [29]. Moreover, association of P16 hypermethylation with cancer progression and lymph node invasion has been shown in different studies [30, 31]. Then, the aberrant methylation signatures of these genes found in the metastatic lymph node can provide a further clue to establish useful biomarkers for screening metastasis in breast cancer. Such biomarkers will need to be additionally explored for the potential opposing functional effects of specific hypermethylated CpG sites on transcriptional activity as well as for absolute percent methylation cutoffs for each breast tumor type that would enable reliable utility of them in the clinical laboratory setting. The possibility of using these hypermethylated genes as biomarkers in our previous study was investigated with the aim of developing a blood based panel for plasma and serum samples of breast cancer patients [18]. Thus the detection of these three hypermethylated genes either as tissue specific biomarkers or as circulating biomarkers may give insight into the prognosis and therapeutic management of the breast cancer patients. Moreover, we identified significantly greater hypermethylation proportion of BMP6 in the lymph node metastasis than their primary tumor tissue (P < 0.05). BMP6 is a member of TGF-β super family and critically involved in many developmental processes [32]. Recently, the close association of BMP6 with progression of tumorigenesis and regulation of invasion for tumor cells has been reported [33]. It has been assumed that demethylation of BMP6 and re-expression of this gene might modulate metastasis and invasion in breast cancer [34, 35].

Pathway analysis of the hypermethylated genes in lymph node metastasis including their biological processes and association to breast carcinogenesis and neoplasm metastasis.
Conclusions
In conclusion, the present study showed aberrant tumor-specific methylation alterations for APC, BIN1, BMP6, BRCA1, CST6, ESR-b, GSTP1, P14, P16, P21, PTEN and TIMP3 in the studied cohort. Additionally, we identified methylation heterogeneity between primary tumors and metastatic lesions. The contribution of aberrant methylation alterations of BMP6, BRCA1 and P16 genes in lymph node metastasis might provide a further clue to establish useful biomarkers for screening metastasis, which might improve prognosis and therapeutic management of the breast cancer patients.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 2009, 59 (4): 225-249. 10.3322/caac.20006.
Lindahl T, Engel G, Ahlgren J, Klaar S, Bjohle J, Lindman H, Andersson J, von Schoultz E, Bergh J: Can axillary dissection be avoided by improved molecular biological diagnosis?. Acta Oncol. 2000, 39 (3): 319-326. 10.1080/028418600750013087.
Reintgen D, Giuliano R, Cox CE: Sentinel Node Biopsy in Breast Cancer: An Overview. Breast J. 2000, 6 (5): 299-305. 10.1046/j.1524-4741.2000.20057.x.
Chambers AF, Groom AC, MacDonald IC: Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002, 2 (8): 563-572. 10.1038/nrc865.
Steeg PS: Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006, 12 (8): 895-904. 10.1038/nm1469.
Pantel K, Brakenhoff RH: Dissecting the metastatic cascade. Nat Rev Cancer. 2004, 4 (6): 448-456. 10.1038/nrc1370.
Baylin S, Bestor TH: Altered methylation patterns in cancer cell genomes: cause or consequence?. Cancer Cell. 2002, 1 (4): 299-305. 10.1016/S1535-6108(02)00061-2.
Esteller M: CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002, 21 (35): 5427-5440. 10.1038/sj.onc.1205600.
Shinozaki M, Hoon DS, Giuliano AE, Hansen NM, Wang HJ, Turner R, Taback B: Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin Cancer Res. 2005, 11 (6): 2156-2162. 10.1158/1078-0432.CCR-04-1810.
Zidan J, Dashkovsky I, Stayerman C, Basher W, Cozacov C, Hadary A: Comparison of HER-2 overexpression in primary breast cancer and metastatic sites and its effect on biological targeting therapy of metastatic disease. Br J Cancer. 2005, 93 (5): 552-556. 10.1038/sj.bjc.6602738.
Feng W, Orlandi R, Zhao N, Carcangiu ML, Tagliabue E, Xu J, Bast RC: Yu Y: Tumor suppressor genes are frequently methylated in lymph node metastases of breast cancers. BMC Cancer. 2010, 10 ( ): 378-
Chen L, Zhu Z, Sun X, Dong XY, Wei J, Gu F, Sun YL, Zhou J, Dong JT, Fu L: Down-regulation of tumor suppressor gene FEZ1/LZTS1 in breast carcinoma involves promoter methylation and associates with metastasis. Breast Cancer Res Treat. 2009, 116 (3): 471-478. 10.1007/s10549-008-0147-6.
Cavalli LR, Urban CA, Dai D, de Assis S, Tavares DC, Rone JD, Bleggi-Torres LF, Lima RS, Cavalli IJ, Issa JP, et al: Genetic and epigenetic alterations in sentinel lymph nodes metastatic lesions compared to their corresponding primary breast tumors. Cancer Genet Cytogenet. 2003, 146 (1): 33-40. 10.1016/S0165-4608(03)00123-7.
Andrews J, Kennette W, Pilon J, Hodgson A, Tuck AB, Chambers AF, Rodenhiser DI: Multi-platform whole-genome microarray analyses refine the epigenetic signature of breast cancer metastasis with gene expression and copy number. PLoS One. 2010, 5 (1): e8665-10.1371/journal.pone.0008665.
Rodenhiser DI, Andrews J, Kennette W, Sadikovic B, Mendlowitz A, Tuck AB, Chambers AF: Epigenetic mapping and functional analysis in a breast cancer metastasis model using whole-genome promoter tiling microarrays. Breast Cancer Res. 2008, 10 (4): R62-10.1186/bcr2121.
Radpour R, Kohler C, Haghighi MM, Fan AX, Holzgreve W, Zhong XY: Methylation profiles of 22 candidate genes in breast cancer using high-throughput MALDI-TOF mass array. Oncogene. 2009, 28 (33): 2969-2978. 10.1038/onc.2009.149.
Barekati Z, Radpour R, Kohler C, Zhang B, Toniolo P, Lenner P, Lv Q, Zheng H, Zhong XY: Methylation profile of TP53 regulatory pathway and mtDNA alterations in breast cancer patients lacking TP53 mutations. Hum Mol Genet. 2010, 19 (15): 2936-2946. 10.1093/hmg/ddq199.
Radpour R, Barekati Z, Kohler C, Lv Q, Bürki N, Diesch C, Bitzer J, Zheng H, Schmid S, Zhong XY: Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS One. 2011, 6 (1): e16080-10.1371/journal.pone.0016080.
Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, Cantor CR, Field JK, van den Boom D: Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A. 2005, 102 (44): 15785-15790. 10.1073/pnas.0507816102.
Radpour R, Haghighi MM, Fan AX, Torbati PM, Hahn S, Holzgreve W, Zhong XY: High-Throughput Hacking of the Methylation Patterns in Breast Cancer by In vitro Transcription and Thymidine-Specific Cleavage Mass Array on MALDI-TOF Silico-Chip. Mol Cancer Res. 2008, 6 (11): 1702-1709. 10.1158/1541-7786.MCR-08-0262.
Radpour R, Sikora M, Grussenmeyer T, Kohler C, Barekati Z, Holzgreve W, Lefkovits I, Zhong XY: Simultaneous Isolation of DNA, RNA, and Proteins for Genetic, Epigenetic, Transcriptomic, and Proteomic Analysis. J Proteome Res. 2009, 8 (11): 5264-5274. 10.1021/pr900591w.
Metge BJ, Frost AR, King JA, Dyess DL, Welch DR, Samant RS, Shevde LA: Epigenetic silencing contributes to the loss of BRMS1 expression in breast cancer. Clin Exp Metastasis. 2008, 25 (7): 753-763. 10.1007/s10585-008-9187-x.
Mehrotra J, Vali M, McVeigh M, Kominsky SL, Fackler MJ, Lahti-Domenici J, Polyak K, Sacchi N, Garrett-Mayer E, Argani P, et al: Very high frequency of hypermethylated genes in breast cancer metastasis to the bone, brain, and lung. Clin Cancer Res. 2004, 10 (9): 3104-3109. 10.1158/1078-0432.CCR-03-0118.
Rivenbark AG, Livasy CA, Boyd CE, Keppler D, Coleman WB: Methylation-dependent silencing of CST6 in primary human breast tumors and metastatic lesions. Exp Mol Pathol. 2007, 83 (2): 188-197. 10.1016/j.yexmp.2007.03.008.
Radpour R, Barekati Z, Haghighi MM, Kohler C, Asadollahi R, Torbati PM, Holzgreve W, Zhong XY: Correlation of telomere length shortening with promoter methylation profile of p16/Rb and p53/p21 pathways in breast cancer. Mod Pathol. 2010, 23 (5): 763-772. 10.1038/modpathol.2009.195.
Kim JH, Jung EJ, Lee HS, Kim MA, Kim WH: Comparative analysis of DNA methylation between primary and metastatic gastric carcinoma. Oncol Rep. 2009, 21 (5): 1251-1259.
Messick CA, Church JM, Liu X, Ting AH, Kalady MF: Stage III colorectal cancer: molecular disparity between primary cancers and lymph node metastases. Ann Surg Oncol. 2010, 17 (2): 425-431. 10.1245/s10434-009-0783-z.
Scheel C, Onder T, Karnoub A, Weinberg RA: Adaptation versus selection: the origins of metastatic behavior. Cancer Res. 2007, 67 (24): 11476-11479. 10.1158/0008-5472.CAN-07-1653. discussion 11479-11480
Feng J, Hu LH, Lu J, Li YR, Xie F: Diagnostic value of BRCA1 and p16 gene methylation in sporadic breast cancer. Ai Zheng. 2009, 28 (4): 436-440.
Zhao YF, Shen SP, Jiang JY, Geng H, Guo JG, Xie LP: Methylation and expression of gene p16INK4a and RB in breast carcinoma. Zhonghua Bing Li Xue Za Zhi. 2010, 39 (6): 377-381.
Su PF, Huang WL, Wu HT, Wu CH, Liu TY, Kao SY: p16(INK4A) promoter hypermethylation is associated with invasiveness and prognosis of oral squamous cell carcinoma in an age-dependent manner. Oral Oncol. 2010, 46 (10): 734-739. 10.1016/j.oraloncology.2010.07.002.
Akiyoshi T, Uchida K, Tateyama S: Expression of bone morphogenetic protein-6 and bone morphogenetic protein receptors in myoepithelial cells of canine mammary gland tumors. Vet Pathol. 2004, 41 (2): 154-163. 10.1354/vp.41-2-154.
Dai J, Keller J, Zhang J, Lu Y, Yao Z, Keller ET: Bone morphogenetic protein-6 promotes osteoblastic prostate cancer bone metastases through a dual mechanism. Cancer Res. 2005, 65 (18): 8274-8285. 10.1158/0008-5472.CAN-05-1891.
Yang S, Du J, Wang Z, Yuan W, Qiao Y, Zhang M, Zhang J, Gao S, Yin J, Sun B, et al: BMP-6 promotes E-cadherin expression through repressing deltaEF1 in breast cancer cells. BMC Cancer. 2007, 7: 211-10.1186/1471-2407-7-211.
Zhang M, Wang Q, Yuan W, Yang S, Wang X, Yan JD, Du J, Yin J, Gao SY, Sun BC, et al: Epigenetic regulation of bone morphogenetic protein-6 gene expression in breast cancer cells. J Steroid Biochem Mol Biol. 2007, 105 (1–5): 91-97.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/12/244/prepub
Acknowledgements
We are indebted to the patients for their cooperation. We thank Ms. Vivian Kiefer for her help. This work was supported by Swiss National Science Foundation (320030_124958/1).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
There is no conflict of interest in the present study.
Authors’ contributions
XYZ and ZB conceived and designed the experiment, ZB carried out experiments, ZB and RR analyzed the data, HZ and QL, provided samples, JB, PL, PT, XYZ material and analysis tools. ZB and XYZ were involved in writing the paper; all authors had final approval of the submitted and published versions.
Electronic supplementary material
12885_2011_3239_MOESM1_ESM.doc
Additional file 1: Supplementary Data 1. The sequence of PCR primers, PCR conditions and complete data for high-throughput methylation analysis of informative CpG sites in 12 breast cancer-related genes, including: gene location, amplicon size and two-way hierarchical cluster analysis are illustrated in dataset. (DOC 204 KB)
12885_2011_3239_MOESM2_ESM.doc
Additional file 2: Supplementary Data 2. High-throughput methylation analysis of CpG sites for 12 candidate genes that are related to breast cancer. (DOC 9 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Barekati, Z., Radpour, R., Lu, Q. et al. Methylation signature of lymph node metastases in breast cancer patients. BMC Cancer 12, 244 (2012). https://doi.org/10.1186/1471-2407-12-244
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-12-244