
Abstract
Background
Autosomal recessive hyper-IgE syndrome (AR-HIES) caused by DOCK8 gene is a rare immunodeficiency disease, the main clinical manifestations include recurrent Eczema-like rash, skin and lung abscesses, accompanied with increased serum IgE level. Here, we report a 7-year-old Chinese girl with a new clinic features caused by DOCK8 gene mutations.
Case presentation
A 7-year-old girl was admitted to our hospital because of abnormal walking posture. The clinical manifestations of the patient included abnormal gait, eczema-like rash, fingertip abscess, high muscle tone, and facial paralysis. Among them, high muscle tone and facial paralysis are new clinic features which have not been reported previously. The blood eosinophils and serum IgE levels were significantly increased, and the lymphocyte subsets indicated a decrease of T lymphocytes. The magnetic resonance imaging (MRI) of her brain suggested myelin dysplasia and brain atrophy. Two novel compound heterozygous mutations (c.1868 + 2 T > C and c.5962-2A > G) of DOCK8 gene were identified by whole exome sequencing. By literature review, there are 11 mutations of DOCK8 gene in Chinese AR-HIES patients.
Conclusions
Two novel splice-site mutations(c.1868 + 2 T > C and c.5962-2A > G) of DOCK8 gene and new clinic features were found in a Chinese girl with AR-HIES, which extends our understanding of DOCK8 gene mutation spectrum and phenotype of AR-HIES in children.
Similar content being viewed by others


Background
Hyper IgE Syndrome (HIES), also known as Job Syndrome, was first reported by Davis in 1966 [1], HIES is a rare immunodeficiency disease, the main clinical manifestations include recurrent Eczema-like rash, skin and lung abscesses, accompanied with increased serum IgE levels. The incidence of this disease is less than 1/100,000, and it usually occurs in infants and young children without gender and ethnic differences [2]. HIES is divided into autosomal dominant HIES (AD-HIES) and autosomal recessive HIES (AR-HIES) according to different inheritance gene. AD-HIES caused by STAT3 mutation and AR- HIES caused by PGM3, SPINK5, DOCK8 and TKY2 mutations have been reported [3].
The DOCK8 gene was firstly reported by Englharts et al. in 2009 [4], it contains 46–48 exons (transcript: NM_001190458.1 at chr9:273,048–465,259; NM_001193536.1 at chr9:273,048–465,259) spanning 190 kb, and maps chromosome 9p24.3. DOCK8 is a member of the DOCK180 family of atypical guanine nucleotide exchange factors (GEF). The DOCK180 family protein domain is also called DHR, binding domain or CZH binding domain, it can combine with RAC, CDC42 and other families to form a complex structure, remove GDP, and promote GTP to bind and active PhoGTP enzyme. However, the substrate that binds to the DOCK8 molecule is currently unclear [5]. DOCK protein plays an important role in cytoskeletal organization, affecting the migration of dendritic cells. DOCK8 deficiency leads to the continuous existence of germinal center B cells, early T cell apoptosis, and reduces natural killer cell (NK cell) toxicity [5]. The pathogenesis of AR-HIE patients caused by DOCK8 gene mutation is related to cellular and/or humoral immunity. The number of T cells is significantly lower than normal, and may be accompanied by a decrease in memory B cells.
Case presentation
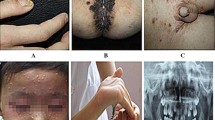
The index patient was a 7-year-old girl from a non-consanguineous family, and she was admitted to our hospital with abnormal walking posture. Her growth and development assessment indicated height of 107 cm(< 3rd) and weight of 20 kg(10th-25th). The patient was able to walk normally at 1 year old, one year later, she appeared abnormal gait, and was prone to wrestling. Therefore, she was misdiagnosed as cerebral palsy, and her symptoms did not improve significantly after rehabilitation. When she was 3 years old, she showed repeated facial eczema, suppuration, and finger abscesses (see Fig. 1 A, B). Physical examination showed eczema-like rashes and abscesses on the face, and the fat under the skin was thin. Her muscle tension of both lower limbs was high, and tension of ankle joint was high. Muscle strength was normal, ankle clonus was positive. Foot inversion, knee extension, and gait instability were observed when walking. Since last year, the patient had 6 times unilateral peripheral facial paralysis, which is a clinic feature not seen in previous reports. Her parents had no clinical symptom as same as the patient. This patient's blood routine results suggested elevated eosinophils, the serum IgE was more than 3200 IU/ml (≤ 90 IU/ml). The results of serum lymphocyte subsets suggested that total T lymphocytes were low and total B lymphocytes were normal; helper T cell/suppressor T cell (Th/Ts) fluctuated between 0.21 and 0.42, which were significantly lower than normal levels (1.03–2.09); the percentage of assisted/induced T lymphocytes fluctuated between 9.26%-14.75%, which were lower than the normal level (18.2%-30.6%); the NK cells ranged from 18.01% to 31.79%, which was higher than the normal level (6.9%-19.3%). Compared with the first admission 2 years ago, the recent MRI of patient suggested that myelin dysplasia and the cerebellar atrophy was aggravated (see Fig. 2), the MRI-DWI of her head was shown in Fig. 3.

A The patient with repeated eczema-like rash on the face. B The patient with repeated suppuration of the fingertips

Comparison of MRI image between the first admission and recent time. A and C MRI on the first admission of the patient. B and D Recent MRI of the patient

The heard MRI-DWI of the patient
There were two splice-site mutations (c.1868 + 2 T > C, c.5962-2A > G) of DOCK8 gene in the patients’ WES (see Fig. 4). His father and mother carried the same heterozygous mutation as the patient, respectively, which were consistent with compound heterozygous mutation. According to ACMG criteria, the two splice-site mutations were classified as possibly pathogenic (PVS1 and PM2).

Sanger sequencing results of the patient and her parents. A chr9:370,302, c.1868 + 2 T > C B chr9:452,009, c.5962-2A > G
Literature review of Chinese patients with DOCK8 gene mutation
To date, more than 130 mutations have been reported in DOCK8 gene all over the world, gross deletions are the most mutations among them. There are 11 different mutations of DOCK8 gene in Chinese patients with autosomal recessive high IgE syndrome (Table 1) [6,7,8,9,10,11]. Among the 11 patients, 3 patients had atopic dermatitis, 8 patients had eczema, 10 patients had a certain degree of infection and allergy symptoms. Among these 10 infected patients, 3 patients had repeated respiratory infection, 3 patients had sinusitis, 8 patients had otitis media, 5 patients had stomatitis, 2 patients had conjunctivitis. For pathogens, including herpes simplex virus (HSV), human papilloma virus (HPV), cytomegalovirus, herpes simplex virus, measles virus, candida, hepatitis B virus. It is worth noting that none of the 11 patients had neurological symptoms. Now we found a compound heterozygous mutation (c.1868 + 2 T > C, c.5962-2A > G) of DOCK8 gene, which was not reported in previous Chinese cases, in addition, the patient had neurological symptoms.
Discussion and conclusions
The present paper has reported a compound heterozygous mutation (c.1868 + 2 T > C, c.5962-2A > G) of DOCK8 gene in a Chinese family with AR-HIES. The two splice-site mutations and facial paralysis were not reported previously in the patients with DOCK8 gene mutation. Combined with her clinical features, laboratory test results, and next-genetic sequencing results, we consider that she can diagnose high IgE syndrome. Her lower extremity muscle tone became more and more hypertensive, and cerebellar atrophy also worsened. Since only DOCK8 gene mutations were found in WES and CNV, we thought that her new clinic features were caused by DOCK8 gene mutations. At the beginning of the patients’ admission, we have excluded cerebrovascular, in addition, her symptoms could not be explained by other causes other than the DOCK8 gene mutation.
The characteristics of AR-HIES patients caused by DOCK8 gene mutation include the significant increase of serum IgE level and eosinophilia in almost all patients. Serum IgG levels are usually normal or increased, IgM levels are usually reduced or decreased with age, and IgA levels can be increased, decreased, normal [12]. Zhang et al. suggested 90% of patients had lower total T cells and total B cells, all patients had lower CD4 + T cells, 36% had lower B cells, and 60% had lower NK cells in 2009 [13]. For the patient, her serum IgE was more than 3200 IU/ml, eosinophil count increased, the total T cell count was low, and the total B cell count was normal. Common clinical features of AR-HIE caused by DOCK8 gene mutation include allergy, infection, malignant tumors. The patients caused by DOCK8 gene mutation are susceptible to viral infection, which is related to cellular and humoral immunity. On the one hand, memory B cells are greatly reduced, which makes patients to virus infection. On the other hand, the patients may have excessive lymphocyte proliferation and abnormal IgE plasma cell proliferation. The number of T cells of the patients is significantly lower than normal. Defective T cells are related to cellular immunity and easily lead to viral infections in patients with DOCK8 deficiency. The main viral infection etiologies in patients with DOCK8 deficiency include herpes simplex virus, varicella-zoster virus, human papilloma virus and molluscum contagiosum virus.
In addition, a small number of patients may have severe central nervous system lesions, such as hemiplegia, seizures, ischemic infarction, and subarachnoid hemorrhage [6], central nervous system vasculitis combined with stroke [7]. Among the 20 families reported by Engelhardt et al. [14], 10 patients had neurological symptoms, including 4 meningitis, 2 CNS vasculitis, 2 severe neurological disease (PML), and 2 fatal encephalitis. For the novel clinic phenotype of facial paralysis, we considered that it is caused by autoimmune deficiency. In a clinical study of Bell’s palsy in adults, Aviel et al. suggested that there are some alterations in the lymphocyte subsets of the peripheral blood during the acute stage of the disease [15]. Gorodezky et al. suggested that compared with control patients, total T cells (CD3) and T helper/inducer cells (CD4) have been decreased in the acute phase of the disease [16].
Compared with AD-HIES, AR-HIES has no clear diagnostic criteria, according to the clinical characteristics and laboratory tests of patients, genetic testing as early as possible is an important means to diagnose such disease.
The patient was identified two heterozygous mutations in the spite-site of the DOCK8 gene (c.1868 + 2 T > C, c.5962-2A > G). His father and mother carried the same heterozygous mutation as the patient, respectively, which were consistent with autosomal recessive inheritance. The mutations of the patient are located in the region of the splice site, according to the ACMG guidelines, the two mutations are satisfied as PVS1 + PM2. Splice-site mutation may affect the expression of DOCK8 gene and the synthesis of DOCK8 protein, combined with the clinical features, it is considered as pathogenic mutations. There are 11 Chinese patients with AR-HIES caused by DOCK8 mutations in our study. Among them, 10 had large deletions in the exon region, which consistent with common mutation types in HGMD database. There are splicing-site mutations in the DOCK8 gene reported abroad, but there are differences in the clinical phenotype of our patient. Engelhardt et.al reported a patient with splicing-site mutation of DOCK8 gene, her neurological symptoms were meningitis [17]. Patients with AR-HIES caused by DOCK8 gene mutation can be treated with gamma globulin to reduce the incidence of infection, corresponding antiviral treatment can be given to patients with virus infection. Allogeneic hematopoietic stem cell transplantation has achieved relatively definite therapeutic effects in such diseases, and there have been case reports of successful allogeneic hematopoietic stem cell transplantation [18]. In a cohort of 136 people reported by Susanne et al., a study of the overall survival probability of allogeneic hematopoietic stem cell transplantation showed that: 87% at 10 years old, 47% at 20 years old, and 33% at 30 years old [19].
In summary, we report a novel clinical phenotype of the AR-HIES patient caused by DOCK8 gene mutation. Two novel mutations (c.1868 + 2 T > C, c.5962-2A > G) of DOCK8 gene mutation was found in the Chinese patient, which extends our understanding of DOCK8 gene mutation spectrum and phenotype of AR-HIES in children.
Availability of data and materials
All data generated or analysed during the current study are included in this published article.
Abbreviations
- AR-HIES:
-
Autosomal recessive high IgE syndrome
- MRI:
-
Magnetic resonance imaging
- HIES:
-
High IgE Syndrome
- AD-HIES:
-
Autosomal dominant high IgE syndrome
- HSV:
-
Herpes simplex virus
- HPV:
-
Human papilloma virus
References
Davis SD, Schaller J, Wedgwood RJ. Job’s Syndrome Recurrent “cold” staphylococal abscesses. Lancet. 1966;1(7445):1013–5.
Li Chunxiao Yu, Hong. Hyper-IgE Syndrome: recent progress in diagnosis and treatment. Clin Pediatr. 2014;32(1):88–91.
Jing Wu, Hong Li, Chen T-X. Clinical Manifestation of Hyper IgE Syndrome Including Otitis Media. Curr Allergy Asthma Rep. 2018;51:1–7.
Engelhardt KR, McGhee S, Winkler S, et al. Large deletions and point mutations involing the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. Allergy Clin Immunol. 2019;124(6):1289–302.
Sanal O, Jing H, Ozgur T, et al. Additional diverse findings expand the clinical presentation of DOCK8 deficiency. Clin Immunol. 2012;32(4):698–708.
Yingyao He, Bin L, Xuping X. Hyper-IgE syndromes. Clin Otorhinolaryngol Head Neck Sury. 2017;31(11):892–6.
Alkhater SA. CNS vasculitis and stroke as a complication of DOCK8 deficiency: a case report. BMC Neurol. 2016;16:54.
Xue L, Yang Y, Wang S. A novel large deletion of the DOCK8 gene in a Chinese family with autosomal-recessive hyper-IgE syndrome. Eur Acad Dermatol Venereol. 2015;29(3):599–601.
Wang S, Mou W, Xu Z, et al. Autosomal recessive hyper-IgE syndrome in two brother of a Chinese familiy with a novel mutation in DOCK8 gene. Eur Acad Dermatol Venereol. 2018;32(8):302–4.
Qin T, An Y, Liu C, et al. Novel DOCK8 gene mutation lead to absence of protein expression in patients with hyper-IgE syndrome. Immunol Res. 2016;64(1):260–71.
Xuefeng X, Yuanjian S, Xiaofen T, Yanan S, Lanfang T, Zhimin C. New point variation of DOCK8 gene in hyper-IgE syndrome. Chin J Pediatr. 2019;57(3):227–9.
Dimitrova D, Freeman AF. Current status of dedicator of cytokinesis-associated immunodeficiency: DOCK8 and DOCK2. Dermatol Clin. 2017;35(1):11–9.
Zhang Q, Davis JC, Lamborn IT, et al. Combined Immunodeficiency Associated with DOCK8 Mutations. N Engl J Med. 2009;361(21):2046–55.
Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Grimbacher B, Chatila TA. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124:1289–302.
Aviel A, Ostfeld E, Burstein R, Marshak G, Bentwich Z. Peripheral blood T and B lymphocyte subpopulations in Bell's palsy. Ann Otol Rhinol Laryngol. 1983;92:187–91.
Gorodezky C, Carranza JM, Bustamante A, et al. The HLA system and T-cell subsets in Bell's palsy. Acta Otolaryngol. 1991;111:1070–4.
Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez HG, Chen A, Kim HS, Lloret MG, Schulze I, Ehl S, Thiel J, Pfeifer D, Veelken H, Niehuse T, Siepermann K, Weinspach S, Reisli I, Keles S, Genel F, Kutuculer N, Chatila TA. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124:1289–302.
Uygun DFK, Uygun V, Reisli I, et al. Hematopoietic stem cell transplantation from unrelated donors in children with DOCK8 deficiency. Pediatr Transplant. 2017;21(7):1–4.
Aydin SE, Kilic SS, Aytekin C, et al. DOCK8 deficiency: clinical and immunological phenotype and treatment options - a review of 136 patients. Clin Immunol. 2015;35(2):189–98.
Acknowledgements
Not applicable.
Funding
No funding.
Author information
Authors and Affiliations
Contributions
All authors contributed to the intellectual content of this manuscript and approved the final manuscript as submitted. JY drafted the initial manuscript and collected the clinical samples and data. YL interpreted the data and revised the article critically for important intellectual content. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by The Human Ethics Committees of the Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology.
Consent for publication
Written informed consent was obtained from the patient’s parent or guardian for publication of this case report and any accompanying image. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, J., Liu, Y. Autosomal recessive hyper-IgE syndrome caused by DOCK8 gene mutation with new clinical features: a case report. BMC Neurol 21, 288 (2021). https://doi.org/10.1186/s12883-021-02324-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-021-02324-3