
Abstract
Background
Chronic migraine is a neurological condition with a large individual and socioeconomic burden of disease. The recently completed Phase III REsearch Evaluating Migraine Prophylaxis Therapy (PREEMPT) clinical development program established the efficacy and safety of onabotulinumtoxinA as a prophylactic treatment for chronic migraine patients. However, clinical questions remain. A long-term evaluation study of onabotulinumtoxinA aims to address some of the remaining questions in the treatment of chronic migraine. The clinical rationale, study design, and treatment plan of this ongoing study are reviewed in this paper.
Methods/Design
The Chronic migraine OnabotulinuMtoxinA Prolonged Efficacy open Label (COMPEL) study will enroll approximately 500 adult patients with chronic migraine at international sites. Patients will be evaluated over 108 weeks, following a 4-week baseline period. Qualified subjects will receive 155 U of onabotulinumtoxinA every 12 weeks for 9 open-label cycles. The primary endpoint will be mean change from baseline in frequency of headache days at 108 weeks. Other endpoints will include additional assessments of the efficacy and safety of onabotulinumtoxinA and the effect of onabotulinumtoxinA on quality-of-life measures, disability, and health economic outcomes. The impact of onabotulinumtoxinA on common comorbidities (eg, sleep, anxiety, and fatigue) will also be assessed.
Discussion
Recruitment and enrollment are ongoing. Post-approval, open-label studies are often designed to more closely resemble clinical practice and provide an opportunity to continue the evaluation of the efficacy and safety of approved treatments. By creating a large database and analyzing a variety of outcome measures over an extended time frame, the COMPEL study will seek to contribute substantially to the existing knowledge of the chronic migraine population and the long-term management of this debilitating disorder.
Clinical Trial Registration Number
Similar content being viewed by others

Background
Chronic migraine (CM) is a severe neurological disorder with a global prevalence of approximately 2 % of adults [1]. According to the International Headache Society, CM is defined as headache on 15 or more days per month for more than 3 months (at least 8 days should meet criteria for migraine without aura or respond to migraine-specific treatment) [1, 2]. Chronic migraine is less prevalent than episodic migraine (EM; headache on <15 days per month), but it has far-reaching individual and societal impact [1, 2].
Compared with EM patients, patients with CM are more likely to have severe disability and an inability to work, function within society, and manage household chores. Patients with CM report a lower health-related quality of life (HRQoL), greater lost productive time, and greater healthcare resource utilization than EM patients [3–6]. Additionally, psychiatric comorbidities such as depression and anxiety are more prevalent among chronic migraineurs than episodic migraineurs [7]. Other frequent comorbidities include fatigue and sleep disorders [8]. Given the severe burden of illness suffered by CM patients, as well as the comorbidities associated with the condition, it is important to design studies that aim to improve understanding of treatment effects for this debilitating disorder.
Currently, onabotulinumtoxinA is the only therapy specifically approved in the US for prophylaxis of headache in CM patients, and the treatment paradigm and dosing are specific to this product.
The currently understood mechanism of action of onabotulinumtoxinA is via interruption of the inflammatory cascade of events that results in peripheral nociceptor sensitization and, subsequently, central sensitization [9, 10]. With neuronal stimulation, the peripheral sensory nerve ending releases vesicles storing substance P, calcitonin gene-related peptide (CGRP), and other neuropeptides. The fusion of the vesicles with the nerve membrane requires the formation of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex, which then facilitates the release of their cargo by the process of exocytosis [9, 11]. As demonstrated in preclinical studies [12], via receptor-mediated endocytosis, onabotulinumtoxinA enters an intracellular membranous endosome, translocates across the endosome into the cytoplasm and cleaves its SNARE protein target, synaptosomal-associated protein 25 (SNAP-25), which blocks the peripheral release of inflammatory neurotransmitters, which in turn reduces the sensitization of pain nerve fibers [9, 10].
The efficacy and safety of onabotulinumtoxinA in CM was evaluated over a 56-week period (24-week, double-blind period, followed by 32 weeks of open-label use) in the Phase III REsearch Evaluating Migraine Prophylaxis Therapy (PREEMPT) clinical program, the largest study in CM to date [13]. Compared to placebo, onabotulinumtoxinA was more effective in reducing mean frequency of headache days from baseline at every visit from Week 4 to the primary endpoint of Week 24 (−8.4 onabotulinumtoxinA vs. -6.6 placebo; p < 0.001) [13]. Statistically significant improvements from baseline in secondary efficacy variables (eg, frequency of migraine days and moderate or severe headache days, and cumulative hours of headache) were established in patients treated with onabotulinumtoxinA compared to placebo. Patients treated with onabotulinumtoxinA also had a significant reduction in severe (>60) Headache Impact Test-6™ (HIT-6) scores (p < 0.01) and experienced an improvement in HRQoL at all time points, as measured by the Migraine-Specific Quality-of-Life Questionnaire (MSQ) (p < 0.001) [13]. Most adverse events reported in the PREEMPT clinical program were mild to moderate, and all resolved without permanent sequelae [13].
The goal of the Chronic migraine OnabotulinuMtoxinA Prolonged Efficacy open Label (COMPEL) study is to test the following clinical hypotheses: (1) onabotulinumtoxinA is an efficacious form of headache prophylaxis in adults with CM, as measured by a reduction from baseline in the number of headache days over a 28-day period after 108 weeks (9 cycles) of treatment; and (2) onabotulinumtoxinA has an acceptable safety profile over 108 weeks (9 cycles) of treatment. Efficacy and safety measures will be assessed at Week 60 and Week 108. Table 1 summarizes the main objectives of COMPEL. Other exploratory hypotheses will be tested in the COMPEL trial, including whether: (1) onabotulinumtoxinA treatment is associated with improvement in depression (Patient Health Questionnaire [PHQ-9]), sleep (Pittsburgh Sleep Quality Index [PSQI]), anxiety (Generalized Anxiety Disorder Assessment [GAD-7]), and fatigue (Fatigue Severity Scale [FSS]); (2) the benefit of onabotulinumtoxinA treatment is observed in the presence of concomitant prophylactics; (3) onabotulinumtoxinA treatment may be associated with a significant decrease in disability and improvement in HRQoL; and (4) onabotulinumtoxinA treatment may be associated with increased productivity and reduced disability and utilization of healthcare resources by patients.
Methods/design
Study design
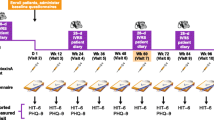
The COMPEL study is a single-arm, open-label, multicenter, post-authorization, prospective study of onabotulinumtoxinA use in CM for headache prophylaxis. The duration of the study is 112 weeks, including a 4-week baseline period and a 108-week, open-label treatment intervention phase (Fig. 1). There will be a total of 11 patient visits, including an initial baseline visit at −4 weeks (Visit 1), 9 interventional visits (on Day 1 and at Weeks 12, 24, 36, 48, 60, 72, 84, and 96), and a study exit visit at Week 108 (Visit 11). Baseline measures of the patient population will include demographics, medical history, physical exam, headache history, detailed headache treatment history, and headache features. Patient diaries will be collected via interactive voice response system (IVRS). Diary data will be entered daily during the 4-week baseline period after Visit 1 and then daily for 28 days, starting 4 weeks prior to Visits 4, 7, 9, and 11.

COMPEL study design. ACM-I = Assessment of Chronic Migraine Impacts; ACM-S = Assessment of Chronic Migraine Symptoms; FSS = Fatigue Severity Scale; GAD-7 = Generalized Anxiety Disorder Assessment; HIT-6 = Headache Impact Test-6; HRU = Healthcare Resource Utilization; IVRS = interactive voice response system; MIDAS = Migraine Disability Assessment Questionnaire; MSQ = Migraine-Specific Quality-of-Life Questionnaire v2.1; PHQ-9 = Patient Health Questionnaire-9; PSQI = Pittsburgh Sleep Quality Index; SF-36 = Short-Form 36 Health Survey; Wk = week
Study eligibility criteria
Study entry screening will be limited to adult patients who are 18 years of age or older, have a diagnosis of CM, are in stable medical condition, are able to follow and comply with study instructions and required visits, and have a negative urine pregnancy test, if applicable. Patients participating in the study may take concurrent medications such as vitamins, herbal remedies, dietary supplements, and a single oral headache prophylaxis medication (ie, beta-blocker, calcium channel blocker, angiotensin-converting enzyme [ACE] inhibitor, aldosterone receptor blocker, antiepileptic, or tricyclic antidepressant), provided these medications are documented. Only one concomitant oral headache prophylaxis medication can be used at any time during the study. A second oral prophylaxis medication may not be added for any patient. All patients on oral headache prophylaxis medication may discontinue this treatment anytime during the study. Patients that discontinue oral prophylaxis may continue treatment only with onabotulinumtoxinA (no alternative oral headache prophylaxis can be added later) (Table 2).
For patients who are taking oral headache prophylaxis medication, patient must be on a stable dose and regimen of a single oral prophylaxis treatment for at least 4 weeks prior to Visit 2 (Day 1). For patients on stable dose and regimen of a single oral headache prophylaxis medication for at least 4 weeks prior to Visit 2, the dose may be changed at or after Visit 4 (Week 24). Those patients who are not taking oral headache prophylaxis at study entry must have discontinued their oral prophylaxis more than 4 weeks prior to Visit 1 to be eligible for inclusion in the study. For patients not on oral headache prophylaxis at Visit 2, a single oral prophylaxis medication such as a beta-blocker, calcium channel blocker, ACE-inhibitor, aldosterone receptor blocker, antiepileptic or tricyclic antidepressant may be added to the patient’s regimen as a component of the headache prevention at or after Visit 4 (Week 24). If a patient is receiving a medication listed as a headache prophylactic that is specifically prescribed for a non‐headache condition, he or she is eligible to enter the study. They are also eligible if receiving another agent on the headache prophylactic list that has been prescribed for headache as long as the prophylactic medication regimen is stable for at least 4 weeks prior to Visit 1.
Patients may take abortive headache medications and may add, change, or decrease these medications as needed if recorded in the patient diary via IVRS. Medications that are both acute and prophylactic treatments will be classified on the basis of the indication for use: if they are specifically prescribed for daily use for headache prevention, they are considered to be prophylactic; if they are used PRN by the patient (even for consecutive days, weeks, or months), they are considered to be acute medications. Patients with previous onabotulinumtoxinA exposure or those with anticipated need for botulinum toxin treatment for any reason during the study (other than study treatment) will be excluded. To continue in the COMPEL study after the 4-week baseline period, participants must have documented ≥15 days of headache per month with headache lasting 4 hours a day or longer. Table 3 summarizes the inclusion and exclusion criteria for COMPEL.
Study treatment and efficacy measures
Patients who meet study criteria will receive 9 onabotulinumtoxinA treatments, administered during Visit 2 (Day 1) through Visit 10 (Week 96). Each treatment will consist of a dose of 155 U of onabotulinumtoxinA administered as 31 fixed-site, fixed-dose intramuscular (IM) injections across 7 specific head and neck muscle areas (Table 4).
The International Headache Society Clinical Trials Guidelines Subcommittee for controlled trials of prophylactic treatment of CM in adults [14] recommends that the primary efficacy measure be based on the number of headache days, reported by patient diaries. The primary outcome in COMPEL is the mean change from baseline in the frequency of headache days for the 28-day period ending at Week 108 (following 9 treatments). Secondary efficacy measures will include both mean change from baseline in the frequency of headache days for the 28-day period ending at Week 60 (following 5 treatments) and mean change from baseline in HIT-6 total score over 4 weeks at Week 108 (following 9 treatments) and at Week 60 (following 5 treatments). The remaining measures described are exploratory efficacy measures. Efficacy measures based on daily diary entries and/or baseline measures will include headache frequency, headache duration (ie, whether headache lasts more than 4 hours), headache severity, and abortive headache medications used. Other measures, based on questionnaires completed at Visit 1 (Screening) through Visit 11 (Week 108), will be as follows: HIT-6, MSQ v2.1, Migraine Disability Assessment Questionnaire (MIDAS), Patient Global Assessment of Treatment (PGAT), GAD-7, PHQ-9, PSQI, FSS, 36-Item Short Form Survey Instrument (SF-36), and healthcare resource utilization (ie, headache-related visits to a healthcare professional, headache-related visits to emergency room or urgent care, headache-related admissions/overnights to a hospital, and headache-related diagnostics in past 6 months) [15–21]. Table 5 includes a short description of each measure that will be collected and assessed in COMPEL in addition to headache days.
Safety and tolerability measures
A key secondary objective of COMPEL is to evaluate the long-term safety and tolerability (9 treatment cycles) of onabotulinumtoxinA for CM in adult patients. Safety and tolerability will be assessed by the frequency and nature of adverse events (AEs) and physical examinations including vital signs. Spontaneous AEs will be analyzed starting on Day 1 (Visit 2), after injection; analysis will continue through the end of the study period in all patients who receive at least 1 injection of onabotulinumtoxinA. Any patient who exhibits change from baseline in the response to Item 9 of the PHQ-9 (“Thoughts that you would be better off dead, or hurting yourself in some way”) any time during the study will consult with the principal investigator about the suicidal ideation and be withdrawn from the study. Patients who are withdrawn from the study will not receive further protocol-related injections and will be referred to appropriate medical care and follow-up.
If a patient becomes pregnant at any time during the study, she will be withdrawn. Specifically, if a female of childbearing potential becomes pregnant during the study, the patient will not receive any further injections and will be followed for 12 weeks after the last study treatment and at least 4 weeks since the pregnancy was reported, before being exited from the study. The investigator will (1) notify the patient’s physician that the patient was being treated with an investigational drug onabotulinumtoxinA, and (2) follow the progress of the pregnancy until delivery and document the outcome of the pregnancy. Patients who are withdrawn from the study will not receive further protocol-related injections and will be referred to appropriate medical care and follow-up.
Statistical considerations: sample size and populations
Up to 40 investigative sites in the US, Australia, and South Korea will enroll a sample size (N = 500 patients) that will provide 80 % power to detect subgroup differences of 2.5 or more headache days as the primary endpoint with a 95 % significance level. The safety population (SP) will include all patients who receive at least 1 onabotulinumtoxinA treatment injection.
Ethical considerations
This study will be conducted in accordance with Institutional Review Board (IRB) regulations (US 21 Code of Federal Regulations [CFR] Part 56.103) or applicable Independent Ethics Committee (IEC) regulations. This protocol is to be conducted in accordance with the applicable Good Clinical Practice (GCP) regulations and guidelines. Written and informed consent will be obtained from each patient or patient’s legally authorized representative prior to enrollment into the study. The Central IRB for the COMPEL Study was reviewed and approved by the Quorum Review IRB in Seattle, WA.
Discussion
As evidenced by several population-based studies, CM is a debilitating disease with extensive individual and socioeconomic burden [3–5], yet only 33 % of CM patients report taking prophylactic medications [3]. Aside from onabotulinumtoxinA, there are no currently approved prophylactic therapy options specifically for CM. There are four botulinum toxin products approved in the US for other indications; three are serotype A (onabotulinumtoxinA [BOTOX®, Allergan, Inc., Irvine, CA, USA], abobotulinumtoxinA [Dysport®, Ipsen, Paris, France], and incobotulinumtoxinA [Xeomin®, Merz Pharmaceuticals GmbH, Frankfurt, Germany]); and one is serotype B (rimabotulinumtoxinB [Myobloc®/Neurobloc®, Solstice Neuroscience, San Francisco, CA, USA]). Each differs in molecular structure, formulation, and clinical profile. There is no international potency reference standard for botulinum toxins, and each formulation of botulinum toxin is different. Therefore, the units of activity are specific to each product and not interchangeable with those of any other botulinum toxins [22].
Several pharmacological agents have been evaluated in small (≤200 patients) double-blind, placebo-controlled studies for prophylaxis of CM [23–27]. In addition to studying a small population, these were generally short-term trials, with the longest study lasting approximately 7 months [28]. Table 6 presents an overview of the clinical trials that have been conducted in a CM population.
The PREEMPT clinical program utilized a set of clearly defined diagnostic criteria and outcome measures to assess the safety and efficacy of onabotulinumtoxinA in the prophylaxis of headache in CM patients. PREEMPT consists of the 2 longest CM trials to date (a 24-week, double-blind period followed by a 32-week, open-label phase) [29, 30]. However, this study did not evaluate the effects of onabotulinumtoxinA treatment past 56 weeks [31]. Consequently, the 108-week COMPEL study aims to assess the efficacy, safety, and tolerability of onabotulinumtoxinA as long-term headache prophylaxis in adult patients with CM.
To our knowledge, COMPEL will be the longest CM trial ever conducted and is expected to add significant knowledge to the literature about long-term use of prophylactic therapy in this patient population. At 15 months, patients who received onabotulinumtoxinA in the PREEMPT study continued to show divergence from those who received placebo during the double-blind phase (Fig. 2); COMPEL will evaluate whether there is continued improvement with onabotulinumtoxinA treatment through 108 weeks, and may or may not demonstrate when a plateau is established. Further, detailed information about the effect of onabotulinumtoxinA on measures other than the primary headache variables will be determined in COMPEL.

PREEMPT 56-week data showed continued improvement in headache days with onabotulinumtoxinA. Source: [31]. This is a pooled analysis of PREEMPT 1 and 2: the double-blind phase included 688 subjects in the onabotulinumtoxinA group and 696 in the placebo group. Headache days at baseline: 19.9 onabotulinumtoxinA group vs 19.8 placebo group, p = 0.498. PREEMPT = Phase 3 REsearch Evaluating Migraine Prophylaxis Therapy
Although open-label studies augment knowledge learned from phase 3 randomized controlled trials, they have inherent limitations. One limitation of our study involves its nonrandomized, open-label design. In addition, an open-label design with long-term follow up can be subject to multiple confounders such as unintentional bias, low persistency rates, and concomitant medication changes. Although steps have been taken to control for concomitant medications and headache medication use, stabilization of dose may not always be possible in patients once enrolled. Finally, COMPEL will capture comorbidities in this patient population; however, not all comorbidities will be captured and patients with severe depressive symptoms and suicidal ideation will be excluded.
In conclusion, COMPEL may provide important clinical guidance for physicians by studying treatment outcomes with onabotulinumtoxinA in the presence of oral prophylactics and measuring the effect of onabotulinumtoxinA on a number of exploratory measures, including the assessment of chronic disease comorbidities such as sleep, fatigue, and anxiety. Healthcare resource utilization will be measured, and an extensive number of patient-reported outcomes will be assessed. Through multiple measures, COMPEL may contribute data to improve our understanding of the long-term effects of onabotulinumtoxinA treatment on CM patients.
References
Natoli J, Manack A, Dean B, Butler Q, Turkel C, Stovner L, et al. Global prevalence of chronic migraine: a systematic review. Cephalalgia. 2010;30:599–609.
Olesen J. Headache Classification Committee of the International Headache Society (IHS): The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33:629–808.
Bigal ME, Serrano D, Reed M, Lipton RB. Chronic migraine in the population: burden, diagnosis, and satisfaction with treatment. Neurology. 2008;71:559–66.
Munakata J, Hazard E, Serrano D, Klingman D, Rupnow MF, Tierce J, et al. Economic burden of transformed migraine: Results from the American Migraine Prevalence and Prevention (AMPP) study. Headache. 2009;49:498–508.
Blumenfeld A, Varon S, Wilcox TK, Buse D, Kawata AK, Manack A, et al. Disability, HRQoL and resource use among chronic and episodic migraineurs: Results from the International Burden of Migraine Study (IBMS). Cephalalgia. 2011;31:301–15.
Stewart WF, Wood GC, Manack A, Varon SF, Buse DC, Lipton RB. Employment and Work Impact of Chronic Migraine and Episodic Migraine. J Occup Environ Med. 2010;52:8–14.
Buse DC, Manack A, Serrano D, Turkel C, Lipton RB. Sociodemographic and comorbidity profiles of chronic migraine and episodic migraine sufferers. J Neurol Neurosurg Psychiatry. 2010;81:428–32.
Peres MF, Zukerman E, Young WB, Silberstein SD. Fatigue in chronic migraine patients. Cephalalgia. 2002;22:720–4.
Aoki KR. Evidence for antinociceptive activity of botulinum toxin type A in pain management. Headache. 2003;43 Suppl 1:S9–15.
Aoki KR. Review of a proposed mechanism for the antinociceptive action of botulinum toxin type A. Neurotoxicology. 2005;26:785–93.
Hargreaves R. New migraine and pain research. Headache. 2007;47:S26–43.
Aoki KR, Francis J. Updates on the antinociceptive mechanism hypothesis of botulinum toxin A. Parkinsonism Relat Disord. 2011;17 Suppl 1:S28–33.
Dodick DW, Turkel CC, DeGryse R, Aurora SK, Silberstein SD, Lipton RB, et al. OnabotulinumtoxinA for treatment of chronic migraine: Pooled results from the double-blind, randomized, placebo-controlled phases of the PREEMPT clinical program. Headache. 2010;50:921–36.
Silberstein S, Tfelt-Hansen P, Dodick DW, Limmroth V, Lipton RB, Pascual J, et al. Guidelines for controlled trials of prophylactic treatment of chronic migraine in adults. Cephalalgia. 2008;28:484–95.
Spitzer RL, Kroenke K, Williams JB, Lowe B. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166:1092–7.
Stewart WF, Lipton RB, Whyte J, Dowson A, Kolodner K, Liberman JN, et al. An international study to assess reliability of the Migraine Disability Assessment (MIDAS) score. Neurology. 1999;53:988–94.
Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606–13.
Buysse DJ, Reynolds III CF, Monk TH, Berman SR, Kupfer DJ. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res. 1989;28:193–213.
Krupp LB, LaRocca NG, Muir-Nash J, Steinberg AD. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch Neurol. 1989;46:1121–3.
Wang SJ, Fuh JL, Lu SR, Juang KD. Quality of life differs among headache diagnoses: analysis of SF-36 survey in 901 headache patients. Pain. 2001;89:285–92.
Jhingran P, Osterhaus JT, Miller DW, Lee JT, Kirchdoerfer L. Development and validation of the Migraine-Specific Quality of Life Questionnaire. Headache. 1998;38:295–302.
Albanese A. Terminology for preparations of botulinum neurotoxins: what a difference a name makes. JAMA. 2011;305:89–90.
Saper JR, Lake III AE, Cantrell DT, Winner PK, White JR. Chronic daily headache prophylaxis with tizanidine: a double-blind, placebo-controlled, multicenter outcome study. Headache. 2002;42:470–82.
Saper JR, Silberstein SD, Lake III AE, Winters ME. Double-blind trial of fluoxetine: chronic daily headache and migraine. Headache. 1994;34:497–502.
Yurekli VA, Akhan G, Kutluhan S, Uzar E, Koyuncuoglu HR, Gultekin F. The effect of sodium valproate on chronic daily headache and its subgroups. J Headache Pain. 2008;9:37–41.
Spira PJ, Beran RG. Gabapentin in the prophylaxis of chronic daily headache: a randomized, placebo-controlled study. Neurology. 2003;61:1753–9.
Silvestrini M, Bartolini M, Coccia M, Baruffaldi R, Taffi R, Provinciali L. Topiramate in the treatment of chronic migraine. Cephalalgia. 2003;23:820–4.
Silberstein SD, Lipton RB, Dodick DW, Freitag FG, Ramadan N, Mathew N, et al. Efficacy and safety of topiramate for the treatment of chronic migraine: a randomized, double-blind, placebo-controlled trial. Headache. 2007;47:170–80.
Diener HC, Dodick DW, Aurora SK, Turkel CC, DeGryse R, Lipton RB, et al. OnabotulinumtoxinA for treatment of chronic migraine: Results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 2 trial. Cephalalgia. 2010;30:804–14.
Aurora SK, Dodick DW, Turkel CC, DeGryse R, Silberstein SD, Lipton RB, et al. OnabotulinumtoxinA for treatment of chronic migraine: Results from the double-blind, randomized placebo-controlled phase of the PREEMPT 1 trial. Cephalalgia. 2010;30:793–803.
Aurora SK, Winner P, Freeman MC, Spierings EL, Heiring JO, DeGryse R, et al. OnabotulinumtoxinA for treatment of chronic migraine: Pooled analyses of the 56-week PREEMPT clinical program. Headache. 2011;51:1358–73.
Blumenfeld A, Silberstein SD, Dodick DW, Aurora SK, Turkel CC, Binder WJ. Method of injection of onabotulinumtoxinA for chronic migraine: a safe, well-tolerated, and effective treatment paradigm based on the PREEMPT clinical program. Headache. 2010;50:1406–18.
Kosinski M, Bayliss MS, Bjorner JB, Ware Jr JE, Garber WH, Batenhorst A, et al. A six-item short-form survey for measuring headache impact: the HIT-6. Qual Life Res. 2003;12:963–74.
Stewart WF, Lipton RB, Kolodner K. Migraine disability assessment (MIDAS) score: relation to headache frequency, pain intensity, and headache symptoms. Headache. 2003;43:258–65.
Kroenke K, Spitzer RL. The PHQ-9: A new depression diagnostic and severity measure. Psych Ann. 2002;32:1–7.
Bartolini M, Silvestrini M, Taffi R, Lanciotti C, Luconi R, Capecci M, et al. Efficacy of topiramate and valproate in chronic migraine. Clin Neuropharmacol. 2005;28:277–9.
Diener HC, Bussone G, Van Oene JC, Lahaye M, Schwalen S, Goadsby PJ. Topiramate reduces headache days in chronic migraine: a randomized, double-blind, placebo-controlled study. Cephalalgia. 2007;27:814–23.
Krymchantowski AV, Silva MT, Barbosa JS, Alves LA. Amitriptyline versus amitriptyline combined with fluoxetine in the preventative treatment of transformed migraine: a double-blind study. Headache. 2002;42:510–4.
Mathew NT, Jaffri SF. A double-blind comparison of onabotulinumtoxinA (BOTOX®) and topiramate (TOPAMAX®) for the prophylactic treatment of chronic migraine: a pilot study. Headache. 2009;49:1466–78.
Cady RK, Schreiber CP, Porter JA, Blumenfeld AM, Farmer KU. A Multi-Center Double-Blind Pilot Comparison of OnabotulinumtoxinA and Topiramate for the Prophylactic Treatment of Chronic Migraine. Headache. 2011;51:21–32.
Acknowledgements
The authors would like to acknowledge Mitchell Brin, MD, for his contributions to this manuscript. The authors also thank Allergan, Inc., for funding Imprint Publication Science, New York, NY, to provide editorial support in the preparation and styling of this manuscript.
Financial Support
This study is being sponsored by Allergan, Inc.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
Dr. Blumenfeld has received honoraria from Allergan, Inc., for consulting and speaking.
Dr. Aurora has received grant/research support and honoraria from Allergan, Inc., for consulting and speaking
Dr. Laranjo was an employee of and held stock in Allergan, Inc.
Dr. Papapetropoulos was an employee of and held stock in Allergan, Inc.
Authors’ contributions
All authors listed on the paper have contributed substantially to the manuscript. AMB, SKA, KL and SP determined the concept and content of this article. AMB and SKA coordinated their efforts on the first draft, and KL and SP critically revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Andrew M. Blumenfeld and Sheena K. Aurora equally contributing authors
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Blumenfeld, A.M., Aurora, S.K., Laranjo, K. et al. Unmet clinical needs in chronic migraine: Rationale for study and design of COMPEL, an open-label, multicenter study of the long-term efficacy, safety, and tolerability of onabotulinumtoxinA for headache prophylaxis in adults with chronic migraine. BMC Neurol 15, 100 (2015). https://doi.org/10.1186/s12883-015-0353-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-015-0353-x