
Abstract
Background
Intellectual disability (ID) constitutes the most common group of neurodevelopmental disorders. Exome sequencing has enabled the discovery of genetic mutations responsible for a wide range of ID disorders.
Case presentation
In this study, we reported on two male siblings, aged 4 and 2 years, with motor and mental developmental delays and mild dysmorphic facial features. To identify the genetic causes of these symptoms, we employed trio-whole exome sequencing for the proband. We found a novel hemizygous missense variant in the PAK3 gene (c.1112G > A, p.Cys371Tyr), which encodes the p21-activated kinase 3, in the proband, which inherited from mother. The younger brother also has the hemizygous variant, which was confirmed by Sanger sequencing. The variant is located in the kinase domain and was regarded as a likely pathogenic variant in this family.
Conclusion
We diagnosed two male siblings with developmental delays as having a PAK3 likely pathogenic variant. This finding expands the list of PAK3 gene mutations associated with neurodevelopmental disorders and provides further details on its clinical features.
Similar content being viewed by others

Background
Intellectual disability (ID) is the terminology used to describe the significant impairment of cognitive and adaptive development due to abnormalities in brain structure or function. ID is a common clinical feature in pediatrics. Early identification and intervention are essential for affected patients. Genetic causes of ID are present in 25–50% of clinical cases [1]. The extensive use of next-generation sequencing (NGS) has resulted in the identification of genetic findings in increasing numbers of patients with developmental disorders [2].
The PAK3 (P21 Activated Kinase 3) gene, which encodes a protein containing a kinase domain (KD) and a p21-binding domain/autoinhibitory domain (PBD/AID), is associated with X-linked non-specific intellectual disability and is known to cause non-syndromic mental retardation X-linked 30/47 (MIM 300558). In the Pak3-knockout mice, oligodendrocyte differentiation in the corpus callosum and the process of synaptic plasticity were affected, the mice showed learning and memory deficiency [3, 4].
To date, there have been 15 families comprising 47 patients reported with PAK3 mutations. The patients showed variable face features and psychiatric manifestations, and all had mild to severe ID. In this study, we identified one novel hemizygous missensevariant of PAK3 in two siblings in one family. We also reviewed the previous cases of PAK3 defects and summarized the phenotype and genotype correlation.
Case presentation
Clinical presentation and family history
The proband was a 3-year-10-month-old boy at the time of admission (Fig. 1A). He was born to a nonconsanguineous couple by normal delivery at 42+ 2 weeks. His birth weight was 3200 g (− 0.2SD), and his Apgar score was 10. He did not meet the developmental milestones and was therefore brought to visit a doctor. He raised his head at 3 months, turned over at 6+ months, and sat up at 9 months. He was able to grab a toy with his hands by himself at 1 year old. He could crawl on his hands and legs and walk independently at 2 years old. He could pinch things with thumb and index finger at 3 years of age but had difficulty putting down objects or releasing them from his fingers. He recognized people at 2.5 years, and could say a single word at 3-year-10-month-old. In the last follow up, he could say a simple sentence at 4-year-2-month-old but could not read or write. However, the acquired skills were not retrogressed. He liked to push people and seemed aggressive. He did not present with seizures but rubbed his legs with his hands while clenching his teeth and demonstrating esotropia for approximately 1 min. This behavior could be interrupted. A 24-h sleep electroencephalogram (EEG) showed little abnormality at 4 years old. Head magnetic resonance imaging (MRI) showed lateral ventricle enlarged, white matter around the lateral ventricle decreased, and with corpus callosum dysplasia (Fig. 1B). His head circumference was 48.6 cm (− 1.2SD) at 4-year-2-month-old. The boy had mild dysmorphic facial feature (hypertelorism, slight epicanthus, esotropia, mild nasal bridge depression, oral hypotonia, open mouth, high palatal arch, large ears). He had a rash in infancy, and at the last follow-up he had psoriasis on the head, back, and shins and depigmentation spots on the forehead. His auditory and visual responses were normal.

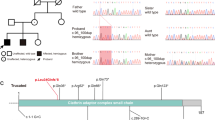
The novel likely pathogenic variant of the PAK3 gene and the brain MRI scans of the patients. a The family tree. b The left sides show sagittal T1 FLAIR images with widened lateral ventricles and thin corpus callosum. The right sides show axial T2 FLAIR images with enlarged lateral ventricles. c The hemizygous variant detected by whole-exome sequencing and confirmed by Sanger sequencing. d The variant region is conserved among human, rhesus, mouse, dog, elephant, chicken and zebrafish. e The predicted structure of the mutated PAK3, with the site of the mutation in the enlarged views
The younger brother was a 2-year-1-month-old boy who was delivered at 42 weeks. His birth weight was 3800 g (+ 0.4SD). He raised his head at 3 months, turned over at 5 months, and sat up at 14 months. He could hold something with his hands by himself at 1 year old and could pinch something with the thumb and index finger until 1.5 years old. In addition, he understood commands at 1.5 years old. He could crawl on his stomach at 23 months, but he still could not crawl with his hands and legs or walk independently. He cannot build blocks, read or write but did not otherwise retrogress. He did not show any abnormal behaviors. The EEG was normal. The head MRI showed lateral ventricle enlarged, white matter around lateral ventricle decreased, and with corpus callosum dysplasia (Fig. 1B). His head circumference was 46.1 cm at 1-year old (− 1.6SD) which was relative small. He also had mild dysmorphic facial features, including hypertelorism, slight epicanthus, esotropia, nasal bridge depression, oral hypotonia, open mouth, high palatal arch, and large ears. He had rash when in infancy and had depigmentation spots on the forehead. Psoriasis appeared in his torso. His auditory and visual sense was normal.
The father and grandparents were healthy. The mother graduated from junior high school with slight difficulty; she had mild facial features, including open mouth and esotropia. The physician and her husband opined that she had a relatively rigid character, which indicating the mother have a mild phenotype.
Genomic analysis
Genomic DNA of the children and their parents was extracted from whole blood using a QIAamp DNA Blood Mini Kit (Catalog no. 51106). Nucleic acid preparation and high-throughput sequencing were performed according to standard protocols in the Clinical Laboratory Improvement Amendments (CLIA) compliant sequencing laboratory in Wuxi NEXTCODE (288 Fute Zhong Road, Waigaoqiao Free Trade Zone Shanghai 200,131, China CLIA ID 99D2064856). Whole-exome capture was performed using an Agilent SureSelect Human All Exon 50 Mb Kit (Agilent Technologies, Santa Clara, California, USA) followed by sequencing as 150-bp paired-end runs on an Illumina XTen (Illumina, San Diego, CA, USA) platform. Segregation of the PAK3 variant within the family was confirmed by Sanger sequencing on the ABI 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Sequence data were mapped to the human reference genome (GRCh37/hg19). Variants calling was using the Genome Analysis Toolkit Best Practices Pipeline (Version 3.2.2) [5]. Data filtering, variant prediction, and interpretation followed ACMG guidelines and those from our previous work [6, 7]. Variant analysis was based on the ExAC database (http://exac.broadinstitute.org/), the 1000 Genomes database (http://www.internationalgenome.org/) and an internal database (more than 30 k samples). The variants were predicted by the online software platforms PolyPhen2.2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), and MutationTaster (http://www.mutationtaster.org/). The structures of PAK3 and its mutant were analyzed using SWISSMODEL (https://www.swissmodel.expasy.org) base on the protein data bank (PDB ID: 6FD3). And the stability results of the wildtype and variant form of protein are reported by MOE.
The mean depth of the trio-whole exome sequencing data was approximately 120×. We identified a novel variant (NM_ 002578: c.1112G > A, p.Cys371Tyr) of PAK3 in the proband, which inherited from mother. Sanger sequencing confirmed his younger brother also had this hemizygous variant (Fig. 1C). The residue is conserved among species, including human, rat, mouse, dog, elephant, chicken, and zebrafish (Fig. 1D). The substituted tyrosine has one more benzene ring than cysteine, the senior structure of the variant protein was not obvious changed (Fig. 1E). The stability of the variant and wildtype protein was analysis using the Residue Scan module in MOE, showed a reduction of variant protein by 3.45 kcal/mol (Additional file 1: Table S1). The variant is located in the kinase domain of the protein (Fig. 2). The variant was not reported in the 1KG, the ExAC, the HGMD, or PubMed and is not recorded in our in-house database (approximately 30,000 samples). The potential pathogenicity of this variant was scored 0, 0.997 and 1 by SIFT, PolyPhen2, and MutationTaster, respectively, which means that the variant was classified as “damaged/disease-causing”. We concluded the variant as a likely pathogenic variant according to the ACMG guidelines, based on two affected patients in this family, the PP1 criteria can be upgraded to PM (Table 1). The other variants consisten with the inheritance model in this family were listed in Additional file 1: Table S2.

A schematic diagram of the mutation in PAK3. Boxes in gray/white with numbers are the exons. Square boxes with dotted lines are untranslated regions. The green frame represents the PBD/AID. The blue frame shows the KD. The dark blue strip shows the microdeletion. The red dot indicates the phosphorylation site at Thr421. The arrows point to the known mutation sites. The missense mutations are listed above the diagram, and the truncation mutations are listed below. The bold red arrow indicates the mutation detected in the present study
Discussion and conclusion
Intellectual disability is a clinically homogeneous but genetically heterogeneous disorder. In 1998, Allen et al. first identified a point mutation (c.1255C > T, p.Arg419*) of the PAK3 gene that affected individuals in a multiplex pedigree with X-linked ID. Including this study, 14 PAK3 mutations in 15 families (family 8 and 9 share the same mutation) have been reported, including 11 missense, 1 nonsense, 1 splicing, and 1 frameshift mutation. Twelve of them were located in the KD, and two were in the PBD/AID. Currently, only 1 deletion in exon 6–18 has been reported, which covers the major area of the PBD/AID and all of the KD [2, 8,9,10,11,12,13,14,15,16,17,18,19,20] (Table 2, Fig. 2).
As the gene encoding p21-activated kinase 3, PAK3 is implicated in dendritic spine morphogenesis and is a key regulator of synaptic functions. The Ala365Glu and Arg419* completely abolish kinase activity [21]. These two variants, along with Lys389Asn and Trp446Ser, result in a failure to phosphorylate myelin basic protein when it is stimulated by the constitutively active form of Cdc42 [10]. Patients with these four mutations showed gross and fine motor skill delay, no or poor/inarticulate speech, epileptic seizures in some cases, and aggressiveness, hyperactivity or other psychoses [10, 12, 13, 19] (Table 2). Arg67Cys, located in the PBD/AID, results in a drastically decreased binding of PAK3 to the small GTPase Cdc42 and impaires the subsequent activation of PAK3 by this GTPase [21]. This mutation was identified in six male patients in one family. Patients in this family showed moderate to severe ID but without seizures, stature growth deficiency or abnormal facial features [9]. The mutations in the PBD/AID might decrease the binding of Cdc42 directly and disturb the pathway, while the mutations in the KD might decrease kinase activity. Therefore, mutations in both the KD and PBD/AID are associated with clinical ID through different molecular mechanisms. In addition, splicing (c.276 + 4A > G, p.Gly92Valfs*35) and deletion ex.6-18del both result in the loss of the PBD/AID and KD. Two patients with these mutations showed facial dysmorphism, microcephaly, and mild to severe ID [16, 17]. The novel likely pathogenic variant of Cys371Tyr identified in our study was located in KD. We performed the structural modeling of the variant protein, found no obvious changed from the wildtype protein. However, the stability of the variant protein calculated by MOE showed reduced. In summary, all patients in 16 families with PAK3 mutations/deletions presented with mild to severe ID regardless of the domain in which the mutation was located.
The patient from family 15, a 17.5-year-old boy with a Ser527Gly mutation, showed severe self-injury and abnormal behaviors. He was treated with low-dose L-dopa/carbidopa and 5-hydroxytryptophan, which significantly improved his self-injurious behavior [14]. In this study, the symptoms of the proband were similar to the patient in family 15. Except for the dysmorphic face and ID, the proband also showed some aggression and hyperactivity. We provided the treatment information of patient in family 15 to clinician, and suggested that earlier treatment may improve the boy’s behavior and prevent the occurrence of severe self-injurious behavior.
The mother in this study showed a slight phenotype. Similarly, the female carrier in family 11 with the heterozygous mutation Trp446Ser showed mild ID and mild dysmorphic facial features, exotropia, hyperactivity, and short attention. Additional, one female patient was reported in family 12, the author descripted the patient had microcephaly, global developmental delay, hypotonia, corpus callosum dysplasia, and stereotypic behavior, who carried a de novo variant of Ser447Phe [2]. Therefore, females might have variable clinical features, for which skewed X-inactivation needs consideration in genetic counseling [13].
In conclusion, we first describe a novel missense variant of PAK3 in two Chinese male siblings with ID and dysmorphic facial features. Additionally, trio-WES could be a useful diagnostic method for allowing timely treatment and genetic counseling.
Availability of data and materials
The likely pathogenic variant of the PAK3 gene has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) with the accession number: SCV000986694. The data that support the findings of this study are available on request from the corresponding author. The raw data are not publicly available due to ethical restrictions.
Abbreviations
- ID:
-
intellectual disability
- KD:
-
kinase domain
- NGS:
-
next-generation sequencing
- PBD/AID:
-
p21-binding domain/autoinhibitory domain
References
Kaufman L, Ayub M, Vincent JB. The genetic basis of non-syndromic intellectual disability: a review. J Neurodev Disord. 2010;2(4):182–209.
Deciphering Developmental Disorders S. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519(7542):223–8.
Meng J, Meng Y, Hanna A, Janus C, Jia Z. Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J Neurosci. 2005;25(28):6641–50.
Maglorius Renkilaraj MRL, Baudouin L, Wells CM, Doulazmi M, Wehrle R, Cannaya V, Bachelin C, Barnier JV, Jia Z, Nait Oumesmar B, et al. The intellectual disability protein PAK3 regulates oligodendrocyte precursor cell differentiation. Neurobiol Dis. 2017;98:137–48.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 10 11–33.
Chen X, Gao Y, Yang L, Wu B, Dong X, Liu B, Lu Y, Zhou W, Wang H. Speech and language delay in a patient with WDR4 mutations. Eur J Med Genet. 2018;61:468.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Hertecant J, Komara M, Nagi A, Al-Zaabi O, Fathallah W, Cui H, Yang Y, Eng CM, Al Sorkhy M, Ghattas MA, et al. A de novo mutation in the X-linked PAK3 gene is the underlying cause of intellectual disability and macrocephaly in monozygotic twins. Eur J Med Genet. 2017;60(4):212–6.
Bienvenu T, Des Portes V, McdonellN CA, Zemni R, Couvert P, Ropers HH, Moraine C, Van Bokhoven H, Fryns JP, et al. Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. Am J Med Genet. 2000;93(4):294–8.
Magini P, Pippucci T, Tsai IC, Coppola S, Stellacci E, Bartoletti-Stella A, Turchetti D, Graziano C, Cenacchi G, Neri I, et al. A mutation in PAK3 with a dual molecular effect deregulates the RAS/MAPK pathway and drives an X-linked syndromic phenotype. Hum Mol Genet. 2014;23(13):3607–17.
Muthusamy B, Selvan LDN, Nguyen TT, Manoj J, Stawiski EW, Jaiswal BS, Wang W, Raja R, Ramprasad VL, Gupta R, et al. Next-generation sequencing reveals novel mutations in X-linked intellectual disability. Omics. 2017;21(5):295–303.
Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC, Walsh CA. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet. 1998;20(1):25–30.
Peippo M, Koivisto AM, Sarkamo T, Sipponen M, von Koskull H, Ylisaukko-oja T, Rehnstrom K, Froyen G, Ignatius J, Jarvela I. PAK3 related mental disability: further characterization of the phenotype. Am J Med Genet A. 2007;143A(20):2406–16.
Horvath GA, Tarailo-Graovac M, Bartel T, Race S, Van Allen MI, Blydt-Hansen I, Ross CJ, Wasserman WW, Connolly MB, van Karnebeek CDM. Improvement of self-injury with dopamine and serotonin replacement therapy in a patient with a Hemizygous PAK3 mutation: a new therapeutic strategy for neuropsychiatric features of an intellectual disability syndrome. J Child Neurol. 2018;33(1):106–13.
Iida A, Takano K, Takeshita E, Hatano CA, Hirabayashi S, Inaba Y, Kosugi S, Kamatani Y, Momozawa Y, Kubo M, et al. A novel PAK3 pathogenic variant identified in two siblings from a Japanese family with X-linked intellectual disability: case report and review of the literature. Cold Spring Harb Mol Case Stud. 2019. https://doi.org/10.1101/mcs.a003988.
Rejeb I, Saillour Y, Castelnau L, Julien C, Bienvenu T, Taga P, Chaabouni H, Chelly J, Ben Jemaa L, Bahi-Buisson N. A novel splice mutation in PAK3 gene underlying mental retardation with neuropsychiatric features. Eur J Human Genet. 2008;16(11):1358–63.
Cartwright A, Smith K, Balasubramanian M. Short case report: Xq23 deletion involving PAK3 as a novel cause of developmental delay in a 6-year-old boy. Clin Dysmorphol. 2017;26(1):38–40.
Al-Shamsi A, Hertecant JL, Souid AK, Al-Jasmi FA. Whole exome sequencing diagnosis of inborn errors of metabolism and other disorders in United Arab Emirates. Orphanet J Rare Dis. 2016;11(1):94.
Gedeon AK, Nelson J, Gecz J, Mulley JC. X-linked mild non-syndromic mental retardation with neuropsychiatric problems and the missense mutation A365E in PAK3. Am J Med Genet A. 2003;120A(4):509–17.
McMichael G, Bainbridge MN, Haan E, Corbett M, Gardner A, Thompson S, van Bon BW, van Eyk CL, Broadbent J, Reynolds C, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatry. 2015;20(2):176–82.
Kreis P, Thevenot E, Rousseau V, Boda B, Muller D, Barnier JV. The p21-activated kinase 3 implicated in mental retardation regulates spine morphogenesis through a Cdc42-dependent pathway. J Biol Chem. 2007;282(29):21497–506.
Acknowledgments
We are very grateful to the patient’s families, the clinicians and our genetic laboratory teams for their contributions to this study.
Funding
This study was funded by the National Natural Science Foundation of China (81801462), and the Shanghai Key Laboratory of Birth Defects (13DZ2260600). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YQ drafted this manuscript, HW and WZ revised the manuscript. YL performed the sequencing data. SW described clinical phenotype of patients. BW, a clinical geneticist analysed the data and confirmed phenotype genotype correlation. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Pretest counseling was performed in the clinic. The parents provided written informed consent to participate, for themselves and on behalf of their children. This study was approved by the ethics committees of Children’s Hospital, Fudan University (2016–235).
Consent for publication
The parents provided written informed consent, for themselves and on behalf of their children, for the publication of identifying images or other personal or clinical details of participants that may compromise anonymity.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1 Table S1
. Stability results of PAK3 and variant, produced by Molecular Operating Environment (MOE). Table S2. The summary of variants consistent with the inheritance model other than the PAK3 gene in this family from the trio-WES data.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Qian, Y., Wu, B., Lu, Y. et al. Novel PAK3 gene missense variant associated with two Chinese siblings with intellectual disability: a case report. BMC Med Genet 21, 31 (2020). https://doi.org/10.1186/s12881-020-0957-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-0957-x