
Abstract
Background
Intellectual disability (ID) is characterized by an IQ < 70, which implies below-average intellectual function and a lack of skills necessary for daily living. ID may occur due to multiple causes, such as metabolic, infectious, and chromosomal causes. ID affects approximately 1–3% of the population; however, the cause can be identified in only 25% of clinical patients.
Methods
To find the cause of genetic ID in a family, we performed whole-exome sequencing and Sanger sequencing to confirm the presence of a SETBP1 variant and real-time quantitative polymerase chain reaction to detect SETBP1 expression in the proband and normal controls.
Results
A novel variant, c.942_943insGT (p. Asp316TrpfsTer28), was found in SETBP1. Furthermore, we observed that SETBP1 expression in patients was only 20% that of normal controls (P < 0.05).
Conclusion
A heterozygous variant in SETBP1 associated with ID was found. This report provides further evidence for its genetic basis and support for clinical genetic diagnosis.
Similar content being viewed by others

Background
Intellectual disability (ID) is characterized by an IQ < 70, which may occur due to environmental or genetic causes. Environmental impacts always occur during pregnancy and childbirth. Toxic substances, pathogens, drugs, and injuries can cause ID. One of the most common mechanisms is through the toxic effects of alcohol, which presents as fetal alcohol syndrome. Genetic abnormalities may cause ID by inducing neurodevelopmental defects or neurodegeneration [1]. Several ID-related genes have been identified, such as SETBP1 [5], HGPRT [2], FMR1 [3], and MECP2 [4]. Variants of SETBP1 can cause mental retardation, autosomal dominant 29 (MRD29), also known as SETBP1 disorder, which is a condition that involves mental retardation, speech and language problems, and non-specific facial features [5, 6]. Little is known about the function of the SETBP1 protein except that it can bind with the SET protein in a particular domain [7].
Here, we report three patients from a non-consanguineous family with the same variant in SETBP1 (c.942_943insGT, p.Asp316TrpfsTer28). They exhibited similar syndromes, including mental retardation and speech problems. The heterozygous frameshift variant led to the truncation of the SETBP1 protein. We suspect that this variant was the cause of ID in this family.
Methods
Patient characteristics
The proband was a 27-year-old woman. She was the third child of non-consanguineous parents. She had difficulty communicating and was unable to go out by herself. Her 29-year-old sister had the same symptoms, while her 31-year-old sister was healthy and had normal cognition.
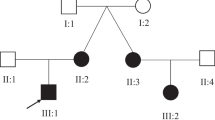
As Fig. 1 shows, the proband’s mother (II-5), sister (III-7), and her sister’s daughter (IV-2) presented the same symptoms. We collected peripheral blood samples from the proband, her mother, and her unaffected elder sister (III-5) and extracted genomic DNA (gDNA). The three individuals provided written approval for their participation in this study and its publication. This study was approved by the Ethics Committee of Central South University, China, and was performed in accordance with the principles of the Helsinki Declaration II.

Clinical features. Genogram. The proband (III-9) is indicated by the arrow
Exome sequencing
Sequencing and analysis were performed as previously described [8]. Samples were submitted to BGI Genomics (Shenzhen, China), which used liquid-capture systems to efficiently capture and enrich human DNA across the entire exome and then provided BGISEQ-500 and HiSeq (Illumina, San Diego, CA) platform services. First, single-stranded circular DNA was replicated through rolling circles to produce DNA nanoballs (DNBs). High-density DNA nanochip technology was used to fix the obtained DNBs on an arrayed silicon chip. A 100-bp paired-end sequence was obtained by combined probe anchor polymerization and double-end sequencing with multiple displacement amplification. Information analysis was initiated with the raw sequencing data. The raw data were filtered to remove adapters, low-quality bases, and undetected bases (represented by N) and compared to the reference genome for single nucleotide polymorphism detection and indel or copy number variation analysis. We then screened harmful sites or genes related to ID according to mutation harmfulness, sample condition, and gene functional phenotype. Thereafter, we used Sanger sequencing to confirm that all patients in this family shared the same SETBP1 variant. Specific PCR primers (SETBP1-F: 5-CACATGGACTGGTCCACCAAC-3 and SETBP1-R: 5-TTTTACTGGACTTTTTCTTGCTGC-3) were used to amplify the target region.
Real-time quantitative polymerase chain reaction
Total RNA of the proband and healthy controls was isolated from peripheral blood using TRIzol (Invitrogen, Carlsbad, CA). Using the GoScript Reverse Transcription System kit (Promega, Madison, WI), we subjected RNA to reverse transcription. The relative mRNA levels of SETBP1 were detected by using real-time quantitative polymerase chain reaction (RT-PCR). Total RNA was converted to cDNA after 40 amplification cycles as follows: 95 °C for 300 s, 95 °C for 10 s, 58 °C for 10 s, and 72 °C for 30 s. The CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA) and MonAmp ChemoHS qPCR Mix (Monad Biotech, Zhuhai, China) were used. GAPDH was used for standardization. The RT-PCR primers are shown in Supplementary Material Table 1.
Statistical analysis
To guarantee the reproducibility of our results, each experiment was performed at least three times, and representative results are shown as mean ± standard deviation. Using the Kruskal‒Wallis test with Steel’s post hoc test, we evaluated differences between the control and proband groups. A difference was regarded as statistically significant at a P value < 0.05.
Results
Case Presentation
The proband (III-9) was a 27-year-old woman with moderate ID. The proband’s mother (II-5), sister (III-7), and her sister’s daughter (IV-2) presented the same symptoms. The proband’s mother (II-5) underwent brain magnetic resonance imaging with normal results. In addition, following primary genetic tests, including the chromosome G-binding test and chromosome microarray analysis (Affymetrix CytoScan 750 K Array) test, the proband did not receive an etiological diagnosis. The clinical information of the patients is presented in the Supplementary Material Table 2.
Gene Identification
We obtained DNA samples from the peripheral blood samples of II-5, III-5, III-9, and III-7 and analyzed these DNA samples through whole-exome sequencing. We found 15 known pathogenic genes that might cause ID.We searched for research reports on the pathogenicity of these genes and found that the phenotype of the patients matched that of MRD29. MRD29, also known as SETBP1 disorder, which is a disease characterized by ID, speech and language problems, and non-specific facial features. In the reported of MRD29, 94% (77/82) of patients had intellectual disabilities, language and motor developmental delays, 47% (37/78) had visual impairments, 53% (17/32) had hearing impairments, and 56% (46/82) had attention/attention deficit issues. A few patients also exhibited other phenotypes (Supplementary Material Table 3). Therefore, we performed Sanger sequencing to validate the SETBP1 mutation in the affected individuals in the pedigree (Fig. 2). Based on the phenotype and variant type of the patients, we identified a SETBP1 variant (c.942_943insGT, p. Asp316TrpfsTer28) as the causative pathogenic variant.

Results of Sanger sequencing. The healthy sister (III-5) had a normal gene sequence. The proband (III-9), proband’s sister (III-7), and proband’s mother (II-5) had the same variant (c.942_943insGT, p. Asp316TrpfsTer28) in SETBP1
SETBP1 expression
To verify the decreased SETBP1 expression in the proband, we extracted mRNA from the proband’s peripheral blood and healthy controls for RT-PCR analysis. The results showed that SETBP1 mRNA levels in the proband were 20% those of her normal sister (III-5) (Fig. 3), indicating that this variant led to a decrease in SETBP1 expression and caused ID.

SETBP1 mRNA expression. SETBP1 mRNA expression in probands was significantly lower than that in controls (P < 0.05)
Discussion
Here, we describe a frameshift variant in SETBP1 in a family with ID, which caused a decrease in SETBP1 expression.
Variants in SETBP1 lead to MRD29 and Schinzel Giedion syndrome (SGS) [5,6,7]. MRD29 is characterized by mild or moderate mental retardation and growth retardation, with most reported variants being frameshift and nonsense variants in SETBP1, while SGS is characterized by growth retardation, special facial features, and multiple deformities, among others, and most reported variants are missense variants in SETBP1 with a typical variant hotspot in the fourth exon. At present, it is suggested that patients with frameshift and nonsense variants in SETBP1 have haploinsufficiency and an MRD29 phenotype, while missense heterozygous variants in hotspots in SETBP1 lead to dominant-negative or gain-of-function effects, and patients develop an SGS phenotype [8,9,10]. Due to the phenotypic heterogeneity of MRD29, there are currently no unified diagnostic criteria. We found that mild intellectual disability, language delay, and delayed motor development were common features among the reported 82 cases. Therefore, by combining whole-exome sequencing to detect SETBP1 mutation types, a more effective diagnosis of MRD29 patients can be made, enabling timely treatment interventions.
SETBP1 is a binding protein that acts as a protein phosphatase 2 A (PP2A) inhibitor by forming a dimer with a SET protein [11, 12]. Ephraim et al. reported that PP2A can inhibit nerve growth in nerve cells. During development, SET-PP2A regulates the differentiation and proliferation of nerve cells, while overexpression or knockout of SET proteins in rat hippocampal neurons can suppress neurites and axon elongation [13]. Therefore, the decreased SETBP1 expression due to the frameshift variant leads to instability of the SETBP1/SET dimer, which cannot inhibit the function of PP2A, thus affecting the development of nerve cells. We consider this a potential molecular mechanism explaining the link between the SETBP1 frameshift variant and mental retardation.
In conclusion, in this family, we found a truncated variant in SETBP1 leading to MRD29. Owing to haploinsufficiency, the patients’ symptoms only included moderate retardation and language delay; for this type of MRD29 patient, whole-exome sequencing is more accurate for diagnosis and can also be helpful for clinical treatment.
Data Availability
The datasets generated for this study can be found in the NCBI Sequence Read Archive (SRA) database (accession number: PRJNA993619), (direct link: https://www.ncbi.nlm.nih.gov/sra/PRJNA993619). Individual-level data can be requested from the corresponding author (hmxiao@csu.edu.cn).
Abbreviations
- DNB:
-
DNA nanoball
- ID:
-
Intellectual disability
- MRD29:
-
Mental retardation, autosomal dominant 29
- RT-PCR:
-
real-time quantitative polymerase chain reaction
- SGS:
-
Schinzel Giedion syndrome
References
SHAFFER L G. American college of medical genetics guideline on the cytogenetic evaluation of the individual with developmental delay or mental retardation[J].Genet Med 2005,7(9):650–4.
SROUR M. SHEVELL M. Genetics and the investigation of developmental delay/intellectual disability[J]. Arch Dis Child 2014,99(1):386–9.
MILLER D T, ADAM M P, ARADHYA S, et al. Consensus statement:microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies[J]. Am J Hum Genet. 2010;86(5):749–64.
WEISE, A,MRASEK K,KLEIN, E et al. Microdeletion and microduplication syndromes[J]. J Histochem Cytochem 2012,60(5):346–58.
Coe BP, Witherspoon K, Rosenfeld JA, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46(10):1063–71.
Filges I, Shimojima K, Okamoto N. Et alReduced expression by SETBP1 haploinsufficiency causes developmental and expressive language delay indicating a phenotype distinct from Schinzel–Giedion syndrome. J Med Genet. 2011;48:117–22.
Liu WL, He ZX, Li F, Ai R, Ma HW. Schinzel-Giedion syndrome: a novel case, review and revised diagnostic criteria. J Genet. 2018;97(1):35–46.
Piazza R, Magistroni V, Redaelli S, et al. SETBP1 induces transcription of a network of development genes by acting as an epigenetic hub. Nat Commun. 2018;9(1):2192.
Leonardi E, Bettella E, Pelizza MF, et al. Identification of SETBP1 mutations by gene panel sequencing in individuals with intellectual disability or with Developmental and Epileptic Encephalopathy. Front Neurol. 2020;11:593446.
Sullivan JA, Stong N, Baugh EH et al. A pathogenic mutationin the SETBP1 hotspot results in a forme-fruste schinzel–giedion syndrome[J]. Am J Med Genet Part A, 2020, 182(8).
Ciccone M, Calin GA, Perrotti D. From the Biology of PP2A to the PADs for therapy of hematologic malignancies. Front Oncol. 2015;5:21.
Cristóbal I, Garcia-Orti L, Cirauqui C, et al. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti-leukemic effect. Leukemia. 2011;25:606–14.
Trakhtenberg EF, Wang Y, Morkin MI, Fernandez SG, Mlacker GM, Shechter JM, Liu X, Patel KH, Lapins A, Yang S, Dombrowski SM, Goldberg JL. Regulating Set-β’s subcellular localization toggles its function between inhibiting and promoting Axon Growth and Regeneration. J Neurosci. 2014;34(21):7361–74.
Acknowledgements
We are grateful to Jiangwan Group for its support of the “Construction Project of the Center of Reproductive Health”.
Funding
This study was supported by the National Key R&D Program of China (2017YFC1001100) and the Construction Project of the Center of Reproductive Health, Central South University (164990007).
Author information
Authors and Affiliations
Contributions
L.W., X.‐D.W., H.-J.T., B.Y., and Y.-Q.P. contributed in the data curation; L.W., X.‐D.W. in the formal analysis; H.‐M.X. in project conceiving, designing, and initiating; L.W., X.‐D.W., X.-M.W. designed and performed the in vitro experiments; H.‐M.X. in supervision; L.W. in writing of the original draft; and L.W., X.‐D.W., B.Y., and H.‐M.X. in writing of the review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All procedures were performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Written informed consent to participate was obtained from all the participants. This study was approved by the Ethics Committee of Central South University, China and was performed in accordance with the principles of the Helsinki Declaration II.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, L., Wang, XD., Yang, B. et al. Novel SETBP1 mutation in a chinese family with intellectual disability. BMC Med Genomics 16, 233 (2023). https://doi.org/10.1186/s12920-023-01649-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01649-x