
Abstract
Background
Mandibulofacial dysostosis with microcephaly (MFDM) is a rare autosomal dominant genetic disease characterized by intellectual and growth retardations, as well as major microcephaly, induced by missense and splice site variants or microdeletions in the EFTUD2 gene.
Case presentation
Here, we investigate the case of a young girl with symptoms of MFDM and a normal karyotype. Whole-exome sequencing of the family was performed to identify genetic alterations responsible for this phenotype. We identified a de novo synonymous variant in the EFTUD2 gene. We demonstrated that this synonymous variant disrupts the donor splice-site in intron 9 resulting in the skipping of exon 9 and a frameshift that leads to a premature stop codon.
Conclusions
We present the first case of MFDM caused by a synonymous variant disrupting the donor splice site, leading to exon skipping.
Similar content being viewed by others


Background
Mandibulofacial dysostosis with microcephaly (MFDM) is a rare autosomal dominant disease characterized by malar and mandibular hypoplasia and microcephaly. Some of its main features include conductive hearing loss, intellectual disability, distinctive facial features and craniofacial malformations that may include characteristic external ear malformations, cleft palate, choanal atresia, and facial asymmetry. In some instances, one observes extracranial malformations such as esophageal atresia (~ 40%), congenital heart disease (~ 40%), and thumb abnormalities (~ 25%). Short stature is present in approximately one-third of individuals [1,2,3,4].
Its exact prevalence is unknown, but more than 80 cases have been described in the literature until now. MFDM is mostly caused by de novo variants in the EFTUD2 gene (MIM# 603892) [5]. In some rarer instances, the MFDM is transmitted from a parent in an autosomal dominant manner (19% of the cases) or due to germline mosaicism (6% of the cases). EFTUD2 encodes the U5-116kD, a highly conserved GTPase component of the major spliceosome complex that processes precursor mRNAs to produce mature mRNAs by allowing the dissociation of U4 and U6 snRNPs during splicing in a GTP-dependent manner [6].
The EFTUD2 gene is composed of 29 exons and presents four transcript variants encoding three different isoforms. Seventy-six distinct single-nucleotide variants (SNVs) and seven microdeletions in EFTUD2 involved in MFDM have been described to date [5]. They can alter basic, surface-forming residues that are potentially available for protein-protein interactions in the internal face of the protein and could conceivably affect protein stability by several mechanisms acting on protein stability, conformation, localization, and/or post-translational modifications. Various types of EFTUD2-variants have been identified, including missense, frameshift, intronic splice site variants and deletions. However synonymous splice site variants in the gene have never been previously implicated in this disease.
Synonymous variants initially do not appear to alter the structure and function of the proteins. They have long been interpreted as “silent” variants. Studies in evolutionary genetics have, however, shown that not all synonymous codons are used at the same frequency in the genome and that selection pressure is exerted even on the synonymous codons as they are used differently for mRNA splicing, translation, and processing machinery. The association of synonymous variants with over 50 human diseases has further confirmed the importance of these phenomena [7].
Case presentation
Here, we report a seven-year-old female patient, who is a native of Libya, who presents postnatal microcephaly to -3SD, sensorineural hearing loss, and global intellectual delay with difficulties of comprehension. She also presents epileptic seizures, livedo and facial dysmorphisms such as micro-retrognatism, malar hypoplasia, dental malocclusion, limitation of mouth opening, and large protruding ears.
As her karyotype was normal and her parents were both healthy, we performed whole-exome sequencing (WES) of the child and her parents to identify putative genetic alterations responsible for this phenotype. WES was performed on genomic DNA prepared from the patient and the parents’ blood samples. The mean coverage of the exome-wide regions was 139.09, 119.25, and 148.62 reads, corresponding to a coverage of at least 10 reads of 95.99, 95.91, and 96.08% of the exome for the patient, mother, and father, respectively. In our variant analysis, we prioritized variants that were rare in the healthy population according to GnomAD v3 database (< 1%), the variants predicted to be deleterious on protein function according to SIFT and PolyPhen tools, and transmitted as compound heterozygous or arose de novo, consistent with the non-consanguineous and healthy parent context (Table S1).
Among these pertinent variants, the only one that could explain the patient’s phenotype was the de novo synonymous variant c.702G > T (transcript NM_004247.4) in the exon 9 of EFTUD2 at position chr17:42956924 (GRCh37/hg19) in the patient (Fig. 1a). This variant replaces a GGG codon to GGT, resulting in the retention of glycine at amino acid residue 234 (p.G234G). According to ACMG 2015 guidelines [8], this variant is classified as having unknown significance. Sanger sequencing confirmed that neither parents carried the variant (Fig. 1b). The variant is located in the G-domain of the protein, which is known to bind and hydrolyze GTP and a site of other variants of EFTUD2 gene that are associated with MFDM (Fig. 1c). As MFDM disease patterns seem to correspond closely to the symptoms of the patient (Table 1), we decided to investigate the potential impact of this synonymous variant on EFTUD2 function.

Identification of the de novo variant in the patient. a The graphs present the Whole Exome Sequencing pile-up reads of the region of interest. The red lines highlight the identified de novo NM_004247.4 c.702G > T variant in EFTUD2 gene in the patient (Top graph) compared to its parent. b Electropherograms from Sanger sequencing of a nucleotide change from G to T in the proband (Daughter) compared to her parents. This variant is a heterozygous variant as both alleles harbor a different nucleotide. c Schematic view of EFTUD2 protein structure, with the location of c.702G > T, p.Gly234Gly and of some other variants of different types causing MFDM disease. The synonymous variant identified in our patient is in red, missense mutations are shown in blue, truncating (nonsense and frameshift) mutations are shown in black, and the intron splice donor mutation is shown in green
The T allele at this position is novel in all public databases, including the NHLBI Exome Sequencing Project, the 1000 Genomes Project, and GnomAD v3, suggesting very high conservation of the G allele in the population. The mutated residue is the last nucleotide of exon 9, localized at the exon/intron junction adjacent to the splice donor site GT (c.702 + 1 and + 2). According to three splicing prediction tools - SpliceSiteFinder-like (SSF), MaxEntScore (MES) and Human Splicing Finder (HSF) - our variant affects the donor splice site by creating an alternative cryptic donor site “GT” preceding the original one (Fig. 2a, b).

In silico predictions of the impact of NM_004247.4 (EFTUD2): c.702G > T variant on splicing. a Screenshot of in silico predictions of the impact of EFTUD2 c.702G > T variant using Alamut software. Three out of five tools predicted that the G > T variant creates a new 5′ donor splicing site (red circle) in the mutated sequence (bottom rectangle) compared to wildtype sequence (top rectangle). The exon 9 (red) and intron 9 (blue) are highlighted on the Figure. b The table represents the splicing scores for the new 5′ donor splicing site by the five predicators tools. The threshold score to reach predicting the presence of a donor splice site is indicated for each predicator tool
To test the prediction, we investigated the consequence of the variant on the splicing of EFTUD2 gene in vivo, in peripheral blood of the proband and her parents. After RNA isolation from leukocytes, we performed an RT-PCR and amplified 360 bases covering exon 8 to exon 12 of EFTUD2 cDNA. We observed in all three individuals the expected PCR product band of ~ 360 bp and an additional PCR product of ~ 280 bp in the proband only (Fig. 3a). This result suggests deletion of about 80 bp in the patient’s EFTUD2 cDNA.

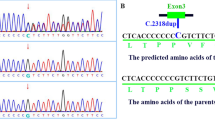
NM_004247.4 (EFTUD2): c.702G > T variant induces exon skipping. a Electrophoresis gel of EFTUD2 cDNA obtained after amplification of the variant region from the proband and its parents. The proband displays two bands, one at 360 bp and one at 280 bp. b Electropherogram from Sanger sequencing 360 bp and 280 bp bands from the electrophoresis gel in A. The exon 8 is highlighted in red, the exon 9 in green and the exon 10 in blue. The exon 9 – exon 10 junction of EFTUD2 cDNA shows exon 9 skipping in cDNA of 280 bp band which is presented only in the proband. c Schematic representation of the exon 9 skipping in the mutant allele of the proband compared to the wild type (WT) allele
The sequencing of the alternative cDNA showed complete deletion of exon 9 (Fig. 3b and c). As the exon 9 length is not a multiple of 3 (83 bp), its deletion would trigger a frameshift leading to a premature stop codon that truncates the protein c.620_702del, p.His209Aspfs*25 (Supplementary Fig. 1). This result demonstrates that the de novo synonymous variant identified in EFTUD2 is responsible for the splicing defect leading to the skipping of exon 9, an exon that is present in all splice isoforms of EFTUD2.
Patients
The patient was recruited at the “Unité de Diagnostic Prénatal - CPDP” of the American Hospital of Paris. The parents gave their signed informed consent for the clinical exome sequencing of their child and themselves.
Whole exome sequencing
Genomic DNA was isolated from peripheral blood using standard protocols. Exome sequencing libraries were prepared with the TruSeq Exome Kit (Illumina, San Diego, CA, USA) following the manufacturer’s recommendations. Paired-end (2 × 75 bp) sequencing was performed on a NextSeq500 sequencer (Illumina, San Diego, CA, USA).
Bioinformatic analysis
FastQ data were aligned to the GRCh37 (hg19) reference genome with bwa-0.7.12 [9], sorted and indexed with samtools-1.2 [10], deduplicated with PICARD-1.110, and base corrected and indel realigned with GATK-3.8 [11, 12]. Variant calling was done with GATK-3.8 HaplotypeCaller in GVCF ERC mode. Variants were called individually for each sample and then combined with GATK-3.8 GenotypeVCFs to produce a combined VCF. The combined VCF was then uploaded and analyzed with Ingenuity Variant Analysis software. Alignments were visualized with GenomeBrowse (Golden Helix - Massachusetts). FastQC-0.11.5 was used to calculate quality metrics for FastQ files and Qualimap-2.2.1 [13] was used to calculate coverage statistics using the truseq-exome-targeted-regions-manifest-v1–2.bed file. The reference file used for alignment and variant calling was human_g1k_v37.fasta which was provided with the GATK b37 resource bundle.
RNA isolation and RT-PCR
Peripheral blood samples from the proband and her parents were used for the analyses in this study. Peripheral blood mononuclear cells were isolated by Ficoll-Paque™ density gradient centrifugation. After total RNA extraction using Trizol, Reverse-Transcription and PCR were performed as described in [14]. Forward and reverse primer sequences purchased from IDT were respectively: 5′ GTGGAATACATGCTTATTAATCCATTGACC 3′ and 5′ GAGCAAGAGAGAGGTGTAGGCATC 3′.
PCR products were analyzed on a 2% agarose gel as described in [14]. Finally, we used PCR clean-up gel extraction from Macherey-Nagel to isolate DNA bands from the agarose gel for sequencing.
Sanger sequencing
The EFTUD2 variant was validated using capillary Sanger sequencing. Briefly, a 262 bp DNA stretch of EFTUD2 was amplified using the Expand Long Template PCR System (Roche, Meylan, France), following the manufacturer’s recommendations. The PCR primer pair was 5′-TTCAAGTTCTCTGGCTCCCA-3′ (forward) and 5′-CCCTCAGTTCACCCTACCAG-3′ (reverse). After purification with the Exostar kit (GE Healthcare, Little Chalfont, UK), PCR products were bi-directionally sequenced with the same primers using Big Dye Terminator Kit v3.1 (Life Technologies). Sequence reactions were run on an ABI PRISM 3730xl sequencer (Life Technologies).
Discussion and conclusions
The increased access to next-generation sequencing for clinical purposes has allowed the identification of thousands of novel pathogenic variants in different individuals. One of the main challenges in clinical genetics is the interpretation of pathogenicity from a sea of variants that remain largely of unknown significance.
Synonymous variants are often interpreted by default as being silent and benign given their predicted null impact on the protein sequence. However, there is evidence for some synonymous SNVs to affect RNA splicing, expression, folding and ultimately function, and, in doing so, contribute to the pathophysiology of many diseases [15,16,17].
In this case study, we report a synonymous c.702G > T variant in the EFTUD2 gene. This variant has not previously been reported in the literature and is absent from large population databases (GnomAD, 1000 Genomes); without further analysis, our initial classification would have been of uncertain significance. However, in silico analysis predicted the disruption of normal splice site, prompting in vitro investigation of its biological significance. The sequencing of the whole exome did not identify other deleterious variants that could be of clinical interest. Although we cannot exclude the presence of relevant deleterious variations in the non-coding regions, the strong correlation between the patient’s phenotype and the clinical consequence of heterozygous alteration of EFTUD2 was sufficient to assume its implication in the disease.
The synonymous variant modifies the consensus sequence between exon 9 and intron 9 from GGG|gt to GGT|gt. In contrary to in silico prediction tools that predicted the creation of an additional GT donor site (Fig. 2), the study of cDNA from blood showed that this variant disrupts the recognition of the donor site by the splicing machinery and results in complete skipping of exon 9. This result could give a hint to the limitations of predictive splicing tools that do not predict the disruption of the splice site induced by this variant. Our study is the first description of synonymous SNV of EFTUD2 in an MFDM patient. Studying cDNA from blood can have some limitations mostly if the gene of interest has different transcripts with a tissue-specific expression; however, we ensured that the EFTUD2 gene is ubiquitously expressed and that the different transcripts do not present differences such as alternative splicing in the region of interest.
Some exonic regions are involved in splicing regulation in highly conserved sites called exonic splice enhancers (ESEs) [18]. In 80% of splicing consensus sites, the last nucleotide of the exon is a “G” which is highly important for the recognition by the splicing machinery [19]. Recently, Savisaar et al. showed that ESEs are under strong selection pressure at synonymous sites, suggesting that synonymous variants in these sites may be a common cause of single-locus genetic diseases [20]. A deleterious missense variant in the last G nucleotide resulting in exon skipping has already been reported in BRCA1 in 2 patients who developed breast cancer at a young age [21] and in a patient with retinitis pigmentosa [22]. To our knowledge, our study is the first to report a deleterious synonymous variant in the final nucleotide of an exon that results in exon skipping.
In conclusion, synonymous variants should not be disregarded especially when they are predicted to affect splicing according to in silico tools. This study provides important evidence for the classification of such variants.
Availability of data and materials
The hg19/GRCh37 human reference genome (https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&position=lastDbPos) was used as the reference dataset in this study. The reference sequence used for the validation of the G234G variant in EFTUD2 was obtained from NCBI Nucleotide using the accession number NM_004247.4. The variant reported in here is available in the Clinvar repository, with accession ID: SCV001251173. The datasets generated during the current study are not publicly available because it is possible that individual privacy could be compromised and the participants did not provide consent to make the data public.
Abbreviations
- ESE:
-
Exonic Splice Enhancer
- EFTUD2:
-
Elongation Factor Tu GTP Binding Domain Containing 2
- HSF:
-
Human Splicing Finder
- MES:
-
MaxEntScore
- MFDM:
-
Mandibulo Facial Dysostosis with Microcephaly
- SNV:
-
Single Nucleotide Variant
- SSF:
-
SpliceSiteFinder-like
- WES:
-
Whole Exome Sequencing
References
Guion-Almeida ML, Zechi-Ceide RM, Vendramini S, Tabith JA. A new syndrome with growth and mental retardation, mandibulofacial dysostosis, microcephaly, and cleft palate. Clin Dysmorphol. 2006;15(3):171–4.
Guion-Almeida ML, Vendramini-Pittoli S, Passos-Bueno MR, Zechi-Ceide RM. Mandibulofacial syndrome with growth and mental retardation, microcephaly, ear anomalies with skin tags, and cleft palate in a mother and her son: autosomal dominant or X-linked syndrome? Am J Med Genet A. 2009;149A(12):2762–4.
Wieczorek D, Shaw-Smith C, Kohlhase J, Schmitt W, Buiting K, Coffey A, et al. Esophageal atresia, hypoplasia of zygomatic complex, microcephaly, cup-shaped ears, congenital heart defect, and mental retardation--new MCA/MR syndrome in two affected sibs and a mildly affected mother? Am J Med Genet A. 2007;143A(11):1135–42.
Wieczorek D, Gener B, Gonzalez MJ, Seland S, Fischer S, Hehr U, et al. Microcephaly, microtia, preauricular tags, choanal atresia and developmental delay in three unrelated patients: a mandibulofacial dysostosis distinct from Treacher Collins syndrome. Am J Med Genet A. 2009;149A(5):837–43.
Huang L, Vanstone MR, Hartley T, Osmond M, Barrowman N, Allanson J, et al. Mandibulofacial Dysostosis with microcephaly: mutation and database update. Hum Mutat. 2016;37(2):148–54.
Hacker I, Sander B, Golas MM, Wolf E, Karagoz E, Kastner B, et al. Localization of Prp8, Brr2, Snu114 and U4/U6 proteins in the yeast tri-snRNP by electron microscopy. Nat Struct Mol Biol. 2008;15(11):1206–12.
Hunt RC, Simhadri VL, Iandoli M, Sauna ZE, Kimchi-Sarfaty C. Exposing synonymous mutations. Trends Genet. 2014;30(7):308–21.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26(5):589–95.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 0 1–33.
Okonechnikov K, Conesa A, Garcia-Alcalde F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 2016;32(2):292–4.
Pasquier J, Galas L, Boulange-Lecomte C, Rioult D, Bultelle F, Magal P, et al. Different modalities of intercellular membrane exchanges mediate cell-to-cell p-glycoprotein transfers in MCF-7 breast cancer cells. J Biol Chem. 2012;287(10):7374–87.
Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3(4):285–98.
Chamary JV, Parmley JL, Hurst LD. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet. 2006;7(2):98–108.
Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 2011;12(10):683–91.
Caceres EF, Hurst LD. The evolution, impact and properties of exonic splice enhancers. Genome Biol. 2013;14(12):R143.
Ma SL, Vega-Warner V, Gillies C, Sampson MG, Kher V, Sethi SK, et al. Whole exome sequencing reveals novel PHEX splice site mutations in patients with Hypophosphatemic rickets. PLoS One. 2015;10(6):e0130729.
Savisaar R, Hurst LD. Exonic splice regulation imposes strong selection at synonymous sites. Genome Res. 2018;28(10):1442–54.
Ladopoulou A, Konstantopoulou I, Armaou S, Efstathiou E, Mihalatos M, Nasioulas G, et al. A change in the last base of BRCA1 exon 23, 5586G-->a, results in abnormal RNA splicing. Cancer Genet Cytogenet. 2002;134(2):175–7.
Demirci FY, Radak AL, Rigatti BW, Mah TS, Gorin MB. A presumed missense mutation of RPGR causes abnormal RNA splicing with exon skipping. Am J Ophthalmol. 2004;138(3):504–5.
Acknowledgements
We are grateful to patients and their families who kindly consented to join the study.
Funding
This study was funded by grants from the Agence Nationale de la Recherche (ANR) (ANR-11-LABX-0070_TRANSPLANTEX), the INSERM (UMR_S 1109), the Institut Universitaire de France (IUF), all the University of Strasbourg (IDEX UNISTRA), the European regional development fund (European Union) INTERREG V program (project n°3.2 TRIDIAG) and MSD-Avenir grant AUTOGEN. No funders participated in the design of the study; the collection, analysis, and interpretation of data; or the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript. AJ, JP, RC, FA and AM performed experimental assays. NH and AJ performed data analysis. AR, SB, PF and KF organized this study, reviewed clinical and laboratory data, and finalized this manuscript. GV determined the initial clinical examination and all clinical tests, reviewed the results of the tests and discussed it with the parents as the referral geneticist. She participated to manuscript writing as well. AJ and JP drafted the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ethics Committee of Weil Cornell Medicine in Qatar approved the study protocol. The ethic committee approval number is 19–00032. Written informed consent was obtained from the parents to take part in this study.
Consent for publication
Written informed consent for publication for identifying images or other personal or clinical details was obtained from both the patient’s parents. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Fig. 1.
mRNA sequence of the WT allele versus the mutant allele. The exon 9 skipping in mutant allele is predicted to cause a frameshift, leading to a premature codon stop. The exon 8 is in red, exon 9 in green and exon 10 in blue.
Additional file 2:
Table S1. Number of prioritized variants during the WES data filtering analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jacob, A., Pasquier, J., Carapito, R. et al. A de novo synonymous variant in EFTUD2 disrupts normal splicing and causes mandibulofacial dysostosis with microcephaly: case report. BMC Med Genet 21, 182 (2020). https://doi.org/10.1186/s12881-020-01121-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01121-y