
Abstract
Background
Wiedemann–Steiner syndrome (WDSTS) is a rare genetic disorder characterized by facial gestalt, neurodevelopmental delay, skeletal anomalies and growth retardation, which is caused by variation of KMT2A gene. To date, only 2 Chinese WDSTS patients have been reported. Here, we report the phenotypes and KMT2A gene variations in 14 unrelated Chinese WDSTS patients and investigate the phenotypic differences between the Chinese and French cohorts.
Methods
Next generation sequencing was performed for each patient, and the variants in the KMT2A gene were validated by Sanger sequencing. The phenotypes of 16 Chinese WDSTS patients were summarized and compared to 33 French patients.
Results
Genetic sequencing identified 13 deleterious de novo KMT2A variants in 14 patients, including 10 truncating, 2 missenses and 1 splicing variants. Of the 13 variants, 11 are novel and two have been reported previously. One of the patients is mosaic in the KMT2A gene. The variation spectra and phenotypic profiles of the Chinese WDSTS patients showed no difference with patients of other ethnicities; however, differ in the frequencies of several clinical features. We demonstrated that variations in the KMT2A gene can lead to both advanced and delayed bone age. We identified 6 novel phenotypes, which include microcephaly, deep palmar crease, external ear deformity, carpal epiphyseal growth retardation, dyslipidemia, and glossoptosis. In addition, patients harbored missense variants in the CXXC zinc finger domain of KMT2A showed more severe neurophenotypes.
Conclusion
Our study consists of the largest cohort of Chinese WDSTS patients that continues to expand the WDSTS phenotypic and variation spectrum. Our results support the notion that the CXXC zinc finger domain of KMT2A gene is a hotspot for missense variants associated with more severe neurophenotypes.
Similar content being viewed by others


Background
Wiedemann–Steiner syndrome (WDSTS, OMIM #605130) is a pleiotropic and extremely rare autosomal dominant disorder, which was first reported by Wiedemann et al. in 1989 and later described by Steiner et al. in 2000 [1]. Using whole-exome sequencing (WES), Jones et al. revealed that germline heterozygous variations of the KMT2A gene (OMIM #159555) was responsible for WDSTS in five patients in 2012 [2]. The KMT2A gene, also known as MLL, encodes a histone lysine methyltransferase that plays a critical role in regulating gene expression during early development and hematopoiesis [3]. Because KMT2A regulates multiple Hox and Wnt genes through histone H3 lysine 4 (H3K4) methylation [4], phenotypes of the WDSTS patients are complex and involve multiple systems, including facial features, skeletal development, and neuro development. A recent summary study pointed out that the clinical features of developmental delay (DD), intellectual disability (ID), postnatal growth retardation, palpebral fissures, down-slanted, wide nasal bridge, broad nasal tip, long eyelashes, and thick eyebrows are more common in WDSTS patients [5]. Increasing number of WDSTS patients of different ethnicities are being identified due to the rapid advance of DNA sequencing technologies, which continues to expand the phenotype spectra with novel features [5]. To date, at least 69 WDSTS patients with variations in KMT2A gene were confirmed, of whom only two are Chinese [5,6,7,8,9,10,11]. Though most of the features in the two Chinese patients are similar to patients of other ethnicities, whether the absent palmar proximal transverse creases is a unique feature of Chinese patients and whether there are novel phenotypes in Chinese patients remain unclear. Meanwhile, our knowledge on the phenotype spectrum and variation spectrum of Chinese patients are still very limited.
Here, we studied the phenotypes and KMT2A variations of 14 unrelated Chinese WDSTS patients. We identified 13 de novo heterozygous variants in the KMT2A gene, including 10 truncating, 2 missense, and 1 splicing variants. Of them, 11 are novel. The phenotypes of Chinese WDSTS patients were compared with a French cohort. In addition, we hereby report several novel clinical features of WDSTS, including macrocephaly, deep palmar crease, external ear deformity, carpal epiphyseal growth retardation, dyslipidemia, and glossoptosis.
Methods
Patients
A total of 14 Chinese patients (8 females and 6 males), aged from 1.5 years to 25 years old, were enrolled from Shanghai Children’s Medical Center, Jiangxi Provincial Children’s Hospital, Shanghai First Maternity and Infant Hospital, Chongqing Children’s Hospital, and Chengdu Women’s and Children’s Central Hospital in China. All patients’ parents are unaffected and non-consanguineous in our study.
Next generation sequencing (NGS)
For Patients 1–7, 10 and 12, proband-only targeted-NGS using inherited disease panel (including 2742 disease-causing genes, cat No.5190–7519, Agilent technologies Inc., Santa Clara, CA, USA) was performed in Shanghai Children’s Medical Center as described previously [12]. For Patients 8, 9, 11 and 14, proband-only targeted NGS was performed by a commercial company (MyGenostics, Inc., Beijing, China), using a clinical exome capture panel containing 4231 disease-causing genes. For patient 13, trio-based WES was performed by the short stature sequencing program [13]. Variants detected by NGS were confirmed by Sanger sequencing in each patient and their parents, when the samples were available.
Statistics analysis
The statistical analysis between two cohorts was performed by Chi-square (χ2) test using the SPSS 17.0 (Statistical Package for the Social Sciences Inc., Chicago, IL, USA). In data statistics, p < 0.05 is considered of suggestive significance.
Results
KMT2A variants
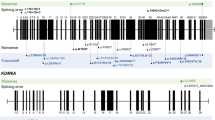
We identified 13 different KMT2A variants (10 truncating, 2 missense, 1 splicing) (Table 1 and Additional file 1: Table S1), with Patient 3 and Patient 7 harboring the same truncating variant (p.Ser774Valfs*12). The two missense variants occurred in the CXXC zinc finger domain (p.Gly1168Asp) and the SET domain (p.Arg3906Cys), respectively (Additional file 2: Table S2). Multiple in silico tools predict deleterious outcomes of these two missense variants (p.Gly1168Asp: scored 0.000 in SIFT, 1.0 in Polyphen-2, 1.0 in MutationTaster, and 35.0 in CADD; p.Arg3906Cys: scored 0.002 in SIFT, 1.0 in Polyphen-2, 1.0 in MutationTaster, and 29.3 in CADD). Of the 13 identified variants, 11 are novel, and the p.Gly1168Asp and p.Ser774Valfs*12 variants have been reported previously [11, 14]. Except the variants identified in patient 9, whose mothers’ sample was not available, the other 12 variants were confirmed to be de novo. According to the variant-interpretation guidelines from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [15], the 2 missense variants were classified as likely pathogenic, and the others are pathogenic. In additional, a total of 106 X of the base at c.5871 in Patient 10 was sequenced. Interestingly, the variant allele (A) has much lower proportion (24 X, 22.6%) when compared to the wild-type allele (T) (82 X, 77.4%), which is confirmed by Sanger sequencing (Additional file 3: Figure S1), indicating mosaicism in the patient.
Clinical presentation
To fully describe the clinical features of Chinese WDSTS patients, 2 previously reported patients [6] were included in our analysis, to make the sample size 16. A comprehensive list of clinical presentation is summarized in Table 1, and Figs. 1 and 2.

Phenotypes of the patients. a Facial appearances of patients 1–14. Patients 10 and 12 showed external ear deformity. b Malformations of hands and feet in seven patients. c Hypertrichosis of seven patients

X-ray results of Patients 6 and 10. a The bone age of Patient 6 was advanced for 2.5 years. Patient 10 had a bone age of 4.5–5 years old and severe carpal epiphyseal growth retardation (b) and scoliosis (c)
In summary, the most frequent (with frequency ≥ 70%) clinical features in Chinese WDSTS patients were as follows: long eyelashes, ID, thick hair, delay in language development, low hair line, hypertelorism, downturned corners of the mouth, down-turned palpebral fissures, vertically narrow palpebral fissures, high palate, hypertrichosis on back, postnatal growth retardation for height (12/16, 75%), and delayed bone age.
Additionally, six clinical features were reported in WDSTS patients for the first time, including macrocephaly in Patient 7, external ear deformity in Patients 10 and 12 (Fig. 1a), deep palmar crease in Patient 9 (Fig. 1b), carpal epiphyseal growth retardation in Patient 10 (Fig. 2b), dyslipidemia in Patient 10 with mild increased in cholesterol level (5.78 mmol/L, normal range: 2.50–5.20 mmol/L) and low-density in lipoprotein level (4.18 mmol/L, normal range: 0–3.36 mmol/L), and glossoptosis in Patient 13.
Intervention
Patient 1 received rehabilitation training to improve the gross motor development before 2 years old. Patient 3 received growth hormone (GH) therapy (0.15 IU/kg/d = 0.05 mg/kg/d) for 14 months and had a 12 cm gain in height (0.85 cm/month).The GH treatment effect of Patient 3 is similar with a recently reported WDSTS patient, whose height velocity increased to 13.3 cm/year [16]. Patient 6 was initially diagnosed with puberty variation with bilateral breast development for the past 5 months at 8-year-10-minth old. She was therefore treated with gonadotrophin releasing hormone (GnRH) analogue to inhibit the rapid sexual development, hoping to improve her adult height. The latest evaluation of Patient 6 at 9-year-4-month old showed well-controlled of the breast development and the normal levels of sex hormones, 17-hydroxyprogesterone, adrenocorticotropic hormone, and dehydroepiandrosterone sulfate. Patient 11 is currently undergoing rehabilitation training to improve her gross motor development. Patient 7 was naturally pregnant with triplets and thus she was provided with genetic counseling. Further examination revealed that one of the three fetuses had scoliosis and one had pulmonary artery atresia. The family decided to take fetal reduction surgery to remove the fetus with scoliosis. A recent follow-up showed that the other two babies were born, and the normal baby was confirmed to carry wild-type KMT2A gene by genetic testing. Unfortunately, we were not able to get genetic information of the baby who had pulmonary artery atresia, since the parents refused to have genetic testing on the baby. Patient 13 had breathing difficulty after birth due to glossoptosis and micrognathia, and thus he underwent a mandibular traction surgery at 50 days of age.
Phenotypic comparison of the Chinese and French cohorts
For further understand WDSTS, we did statistical analysis for the phenotypes between the 16 Chinese patients and a cohort of 33 French patients [11]. As shown in Table 2, no significant differences of age span, gender distribution, and the KMT2A gene variant spectrum in the two cohorts. The overall clinical feature profiles are similar in these two groups, however, several differences still can be observed. Three features of ptosis, thick eyebrows, and feeding difficulties showed significant statistical difference in the two cohorts. The Chinese cohort had higher frequency of ptosis but lower frequency of thick eyebrows and feeding difficulties. In addition, though no statistical difference was observed, postnatal growth retardation, microcephaly, down-turned palpebral fissures, long eyelashes, brachydactyly, and delayed bone age were more frequent in the Chinese cohort, while the frequency of thin upper lip, hyperopia, and congenital heart malformations were higher in the French cohort.
Discussion
In this study, we recruited 14 unrelated Chinese WDSTS patients. To our knowledge, this is the largest cohort of Chinese WDSTS patients with the greatest age span (from pediatric to adult) that have been reported. Recently, Sun et al. reported two Chinese male patients who presented with absent palmar proximal transverse creases, which may be a unique characteristic of Chinese WDSTS patients [6]. We thus examined all the 14 patients and found that no patients had abnormal palmar crease except for Patient 9, who had deep palmar crease (Fig. 1c). We speculate that the lack of abnormal palmar crease in patients of other ethnicities may be a result of negligence during phenotype assessment. Additionally, we revealed several novel clinical features in WDSTS patients, including macrocephaly, deep palmar crease, external ear deformity, carpal epiphyseal growth retardation, dyslipidemia, and glossoptosis.
Though it seems that several clinical features have frequency differences in the Chinese and French cohorts, only 3 (ptosis, thick eyebrows, and feeding difficulties) show statistical significance that might due to lack of enough Chinese WDSTS samples. Prior to the French cohort study, delayed bone age had been reported in at least 3 patients [6, 17], while significantly advanced bone age was first and only described in a 4-year-old female patient [18]. Here, the French cohort and our cohort reported another 9 and 12 patients presented with advanced bone age and delayed bone age, respectively. Therefore, we propose that both of advanced bone age and delayed bone age are authentic phenotype of WDSTS caused by KMT2A variation. Patients 3, 4 and 10 had GH deficiency and also presented with postnatal growth retardation, which are consistent with the results from the French cohort. Moreover, maternity history of Patient 7 supports the surmise of normal fertility in female WDSTS individuals, which has also been reported in two French patients.
The French cohort reported a healthy unaffected father with very low proportion of mosaicism in the KMT2A gene (p.Arg1154Trp), who had two affected daughters. Intriguingly, Patient 10 in our cohort was the first WDSTS case reported with mosaicism in the KMT2A gene (c.5871 T > A; p.Tyr1957*), who on the other hand presented with more severe phenotypes as well as several previously unreported symptoms (external ear deformity, carpal epiphyseal growth retardation, and dyslipidemia).
Because WDSTS can phenotypic overlap with Pierpont syndrome, Cornelia De Lange syndrome and Kabuki syndrome [6, 7, 9, 19, 20], the candidate genes causing the later three syndromes (i.e. TBL1XR1, NIPBL, SMC1A, SMC3, RAD21, HDAC8, KMT2D, and KMD6A) in our cohort have been excluded. To date, a total of 72 KMT2A variants spanning the whole protein have been identified (including the variants identified in this study), of which 71 are point variants and only one is intragenic large deletion [5,6,7,8,9,10,11]. As shown in Additional file 2: Table S2, of the 71 point variants, 48 are truncating, 17 are missense, and 6 are splicing variants. The p.Ser774Valfs*12 variant in Patients 3 and 7 has been previously reported in 3 unrelated patients [11, 21], which qualifies Ser774 as a hotspot for gene variation in KMT2A. Other 5 recurrent variants, including p.Arg1154Trp [11], p.Cys1155Tyr [11, 22], p.Gly1168Asp [14, this study], p.Arg1633* [2, 11] and p.Arg2480* [7, 11] were also identified. Although we did not find hotspot variation region, it is noteworthy that only missense variants (7/17) occurred in the cysteine-rich CXXC zinc finger domain (Additional file 2: Table S2), which is predicted to selectively bind to unmethylated CpG-containing stretches of the target genes [10]. Previous studies suggested that patients who harbor the missense variant in the CXXC DNA binding domain may show more severe neurodevelopmental delay [10, 14]. Indeed, in our cohort, Patient 5 (p.Gly1168Asp) has the most severe anomalies in neurological development. In contrast, Patient 4, who has a missense variant (p.Arg3906Cys) in the SET domain showed less neurological abnormalities, although the SET domain plays an important role in transcriptional activation via its H3K4 methyltransferase activity [5]. In the French cohort, 5 patients (Patients 24–28) harbored the missense variants in the CXXC DNA binding domain. 3 of them showed severe ID, while only one patient with severe ID was observed in the other 28 patients. 4 of them had persistent hypotonia (8 in the other 25 patients) and 3 of them had seizures (only one in the other 26 patients). These results further and firmly demonstrate the relationship between dysfunction in the CXXC DNA binding domain and severe nuerophenotypes.
Most of the KMT2A variants generate truncated products (the same in Chinese patients, 12/16) that further suggest haploinsufficiency is the main pathogenesis of KMT2A gene. It is speculated that missense variants of the KMT2A gene are more likely to cause WDSTS by a dominant negative effect [23]. The hypothesis is supported by the fact that upregulation of the KMT2A gene transcription in patient with p.Arg1154Trp and site-specific DNA methylation changes driven by KMT2A missense variants [9, 10], which might explain the missense variants in the CXXC DNA binding domain linked to a more severe neurophenotypes. Nevertheless, more functional study need be performed to figure out the molecular mechanism, especially why there were only missense variants in this domain.
Currently, it is still difficult to have more conclusions of genotype and phenotype in WDSTS patients due to its broad spectrum of phenotypes. Further studies should attempt elucidating the connection between genotype and phenotype, with epigenotype in consideration. In addition, the clinical interventions for WDSTS patients are yet limited. For most patients, symptomatic treatment, such as rehabilitation training, is the only option to improve the motor development. Results from Patient 3 in our study and another reported 2 patients [6, 16] indicate that GH therapy may be an effective method to improve the height of patients with GH deficiency. Of course, clinical safety should be taken into consideration when using growth hormone therapy.
Conclusions
In summary, we report 14 Chinese WDSTS patients with 13 pathogenic variants in the KMT2A gene, including 11 novel variants. Our detailed comparison between the Chinese and French patients indicates no significant difference in phenotypic spectrum; however, the frequency of several symptoms were different. We demonstrated that variation in the KMT2A gene can lead to both advanced and delayed bone age. The novel phenotypes, including macrocephaly, deep palmar crease, external ear deformity, carpal epiphyseal growth retardation, dyslipidemia, and glossoptosis further extended the WDSTS phenotype spectrum. Our study supports the notion that the CXXC zinc finger domain is a hotspot region for missense variants, which is associated with more severe neurophenotypes. In addition, growth curves and endocrine-related problem need further investigation in the future.
Abbreviations
- DD:
-
Developmental delay
- GH:
-
Growth hormone
- GnRH:
-
Gonadotrophin releasing hormone
- H3K4:
-
Histone H3 lysine 4
- ID:
-
Intellectual disability
- NGS:
-
Next generation sequencing
- WDSTS:
-
Wiedemann-Steiner syndrome
- WES:
-
Whole-exome sequencing
References
Steiner CE, Marques AP. Growth deficiency, mental retardation and unusual facies. Clin Dysmorphol. 2000;9(2):155–6.
Jones WD, Dafou D, McEntagart M, Woollard WJ, Elmslie FV, Holder-Espinasse M, et al. De Novo Mutations in MLL Cause Wiedemann-Steiner Syndrome. Am J Hum Genet. 2012;91(2):358–64.
Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10(5):1107–17.
Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10(5):1119–28.
Enokizono T, Ohto T, Tanaka R, Tanaka M, Suzuki H, Sakai A, et al. Preaxial polydactyly in an individual with Wiedemann-Steiner syndrome caused by a novel nonsense mutation in KMT2A. Am J Med Genet A. 2017;173(10):2821–5.
Sun Y, Hu G, Liu H, Zhang X, Huang Z, Yan H, et al. Further delineation of the phenotype of truncating KMT2A mutations: the extended Wiedemann-Steiner syndrome. Am. J Med Genet A. 2017;173(2):510–4.
Miyake N, Tsurusaki Y, Koshimizu E, Okamoto N, Kosho T, Brown NJ, et al. Delineation of clinical features in Wiedemann–Steiner syndrome caused by KMT2A mutations. Clin Genet. 2016;89(1):115–9.
Popp B, Ekici AB, Thiel CT, Hoyer J, Wiesener A, Kraus C, et al. Exome Pool-Seq in neurodevelopmental disorders. Eur J Hum Genet. 2017;25(12):1364–76.
Sobreira N, Brucato M, Zhang L, Ladd-Acosta C, Ongaco C, Romm J, et al. Patients with a kabuki syndrome phenotype demonstrate DNA methylation abnormalities. Eur J Hum Genet. 2017;25(12):1335–44.
Lebrun N, Giurgea I, Goldenberg A, Dieux A, Afenjar A, Ghoumid J, et al. Molecular and cellular issues of KMT2A variants involved in Wiedemann-Steiner syndrome. Eur J Hum Genet. 2018;26(1):107–16.
Baer S, Afenjar A, Smol T, Piton A, Gérard B, Alembik Y, et al. Wiedemann-Steiner syndrome as a major cause of syndromic intellectual disability: a study of 33 French cases. Clin Genet. 2018;94(1):141–52.
Hu X, Li N, Xu Y, G1 L, Yu T, Yao RE, et al. Proband-only medical exome sequencing as a cost-effective first-tier genetic diagnostic test for patients without prior molecular tests and clinical diagnosis in a developing country: the China experience. Genet Med. 2017. https://doi.org/10.1038/gim.2017.195.
Hu X, Gui B, Su J, Li H, Li N, Yu T, et al. Novel pathogenic ACAN variants in non-syndromic short stature patients. Clin Chim Acta. 2017;469:126–9.
Min Ko J, Cho JS, Yoo Y, Seo J, Choi M, Chae JH, et al. Wiedemann-Steiner syndrome with 2 novel KMT2A mutations. J Child Neurol. 2017;32(2):237–42.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Stoyle G, Banka S, Langley C, Jones EA, Banerjee I. Growth hormone deficiency as a cause for short stature in Wiedemann-Steiner syndrome. Endocrinol Diabetes Metab Case Rep. 2018. https://doi.org/10.1530/EDM-18-0085.
Calvel P, Kusz-Zamelczyk K, Makrythanasis P, Janecki D, Borel C, Conne B, et al. A case of Wiedemann-Steiner syndrome associated with a 46,XY disorder of sexual development and gonadal Dysgenesis. Sex Dev. 2015;9(5):289–95.
Mendelsohn BA, Pronold M, Long R, Smaoui N, Slavotinek AM. Advanced bone age in a girl with Wiedemann–Steiner syndrome and an Exonic deletion in KMT2A (MLL). Am J Med Genet A. 2014;164A(8):2079–83.
Yuan B, Pehlivan D, Karaca E, Patel N, Charng WL, Gambin T, et al. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest. 2015;125(2):636–51.
Parenti I, Teresa-Rodrigo ME, Pozojevic J, Ruiz Gil S, Bader I, Braunholz D, et al. Mutations in chromatin regulators functionally link Cornelia de Lange syndrome and clinically overlapping phenotypes. Hum Genet. 2017;136(3):307–20.
Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, et al. Enhanced utility of familycentered diagnostic exome sequencing with inheritance model–based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17:578–86.
Bramswig NC, Lüdecke HJ, Alanay Y, Albrecht B, Barthelmie A, Boduroglu K, et al. Exome sequencing unravels unexpected differential diagnoses in individuals with the tentative diagnosis of coffin–Siris and Nicolaides–Baraitser syndromes. Hum Genet. 2015;134:553–68.
Stellacci E, Onesimo R, Bruselles A, Pizzi S, Battaglia D, Leoni C, et al. Congenital immunodeficiency in an individual with Wiedemann-Steiner syndrome due to a novel missense mutation in KMT2A. Am J Med Genet A. 2016;170(9):2389–93.
Acknowledgements
We deeply thank each family for their participation in this study.
Funding
This work was supported by the National Natural Science Foundation of China (Grants No. 81472051, No. 81601869, and No.81772303), Project of Shanghai Municipal Science and Technology Commission (Grant No. 15410722800), Project of Shanghai Municipal Education Commission- Gaofeng Clinical Medicine (Grant No. 20152529), and Project of Shanghai Municipal Commission of Health and Family Planning (Grant No. 20154Y0027).
Author information
Authors and Affiliations
Contributions
NL, YRW, and YY participated in molecular genetic studies and writing of the manuscript; HH, SYX, LMS, MC, CS, XRC, YD, GYC, YC, and YFX collected and submitted clinical information; PPW, TTY, and REY participated in molecular genetic studies and performed NGS experiments and Sanger sequencing; YPS directed this work and edited the manuscript; XMW and JW conceived and designed the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All procedures followed were in accordance with the ethical standards of the responsible institutional committee on human experimentation and with the Helsinki Declaration of 1975 (revised in 2000). The protocols were approved by the Institutional Medical Ethics Committee.
Consent for publication
Informed consent was obtained from each patient’s family.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. The variants of KMT2A gene in Chinese WDSTS patients. (DOCX 19 kb)
Additional file 2:
Table S2. Summarize of the KMT2A variants in WDSTS patients (DOCX 19 kb)
Additional file 3:
Figure S1. DNA sequencing results of patient-10. (A) NGS data, (B) Sanger sequencing result. ‘T’ is the wild-type allele and ‘A’ is the variant allele. (TIFF 3994 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, N., Wang, Y., Yang, Y. et al. Description of the molecular and phenotypic spectrum of Wiedemann-Steiner syndrome in Chinese patients. Orphanet J Rare Dis 13, 178 (2018). https://doi.org/10.1186/s13023-018-0909-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-018-0909-0