
Abstract
Background
Xeroderma pigmentosum (XP) is a rare autosomal recessive genodermatosis. There are eight complementation groups of XP (XP-A to G and a variant form). XP-E is one of the least common forms, and XP-E patients are generally not diagnosed until they are adults due to a later onset of skin alterations.
Case presentation
We report a case of a 28-year-old Chinese woman with freckle-like hyperpigmented macules in a sun-exposed area who is prone to develop basal cell carcinomas. A genetic study revealed a novel homozygous c.111_112del deletion in exon 1 of the DDB2 gene. Western blotting analysis revealed that the patient lacked the expression of the wild-type mature DDB2 protein. The proband was first diagnosed with XPE on the basis of clinical findings and genetic testing. Sun protection was recommended, and the patient did not develop any skin cancers during the one-year follow-up.
Conclusions
We identified a novel homozygous deletion in DDB2 gene in Chinese XP-E patients having unique clinical features.
Similar content being viewed by others


Background
Xeroderma pigmentosum (XP) is a rare autosomal recessive genetic disease characterized by increased sensitivity to ultraviolet radiation-induced sunburn, skin pigmentation, skin cancers, ocular disease and neurological degeneration. XP is classified into eight genetic groups: groups A-G and a variant form (XP-V). The genes responsible for XP-A to XP-G and XP-V are XPA, ERCC3, XPC, ERCC2, DDB2, ERCC4, ERCC5, and POLH [1]. It is estimated that the prevalence of XP is 1/1,000,000 in the USA and Europe, and it is more common in Japan, with an estimated of 1/22,000 [2, 3]. Based on the different mutations of the XP gene, there are at least six different clinical symptoms: XP, XP with neurological symptoms, trichothiodystrophy, XP with trichothiodystrophy symptoms, XP with Cockayne syndrome and cerebro-oculo-facio-skeletal syndrome [4].
Severe sunburn on minimal sun exposure is a conspicuous symptom for the diagnosis of XP, but only approximately 60% of all patients show this manifestation [5]. XP is primarily diagnosed in the clinic and confirmed by functional cell-based systems as well as genetic tests. Because of a deficiency in the restoration of DNA damage induced by UV radiation, patients with XP have a 2000-fold increased risk of melanoma and a 10,000-fold increased risk of nonmelanoma skin cancer [6]. Early diagnosis is of utmost importance to initiate strict and consistent sun protection and regular examination of the skin.
XP-E is one of the least common forms of XP. XP-E patients are generally not diagnosed until they are adults due to the lack of sunburn reaction, and they are prone to develop multiple skin cancers, such as basal cell carcinoma, squamous cell carcinoma and malignant melanoma. DNA-binding protein is a heterodimer of DDB1 and DDB2 and is very important for proper DNA damage recognition involved in nucleotide excision repair (NER) [7]. Mutations of the DDB2 gene are responsible for XP-E.
Here, we report an XP-E patient from a Chinese consanguineous family. The patient harbours a novel homozygous mutation, c.111_112del (p.A39Efs*6), of the DDB2 gene. To our knowledge, XP-E has not yet been reported in Chinese people.
Case presentation
One year ago, a 28-year-old Chinese woman came to our department to consult for recurrent ulceration, bleeding and crusted nodules on her nose. She is the first child of healthy consanguineous parents. The parents of this patient were first cousins. Pregnancy and delivery were normal. She has normal mental development. On examination, numerous freckle-like hyperpigmented macules were seen on her face and neck with an ulcerated bleeding nodule on her nose (Fig. 1a). Reticulate hyper- and hypopigmented macules could be seen on her arm (Fig. 1b). The skin throughout her body was dry and could not be relieved by using cream. She denied history of acute sunburn. Her brother, who suffered burns 5 years ago, also had similar clinical characteristics to the proband. An extensive burn scar and a 0.5 cm × 1 cm hyperpigmented nodule were seen on the face of her brother (Fig. 1c). Biopsy results confirmed the diagnosis of basal cell carcinomas of the hyperpigmented nodules on the nose of the proband and her brother.

Clinical findings of the proband and her brother. a. Freckle-like hyperpigmented macules on her face with an ulcerated bleeding nodule on her nose. b. Hyper- and hypopigmented macules on her arm. c. The proband’s brother has similar clinical characteristics
Genetic study
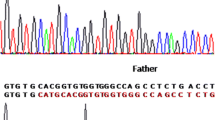
This study was approved by the Ethics Committee of Jinling Hospital and written informed consent was obtained from all participants. Blood samples of the patient and her family members were collected. DNAs were isolated from blood samples. Total mRNA was extracted from the proband’s surgical tissue. cDNA was obtained by reverse transcription. Eight genes responsible for XP were sequenced using next-generation sequencing (NGS), and the result was confirmed by Sanger sequencing. A novel homozygous c.111_112del deletion in exon 1 of the DDB2 gene, which resulted in a frameshift mutation of amino acid p.A39Efs*6, was detected in the proband and her brother (Fig. 2a). Her parents are heterozygous for this mutation (Fig. 2b). Sequencing at the cDNA level also confirmed the mutation (Fig. 2c). This mutation was not found in 120 unrelated, population-matched control individuals. Western blotting analysis revealed that the patient lacked the expression of the wild-type mature DDB2 protein (Fig. 3). The proband and her brother were diagnosed with XPE on the basis of clinical findings and genetic tests. Sun protection was recommended, and the patient did not develop any skin cancers within 1 year of follow-up.

Novel deletion mutation in the DDB2 gene was detected in the patient. a. Homozygous mutation. b. Heterozygous state. c. Confirmation of the mutation at the cDNA level

Western blotting analysis revealed that the patient lacked the expression of the wild-type mature DDB2 protein
Discussion and conclusions
Three pathways in mammalian cells are involved in protection from UV irradiation: global genomic repair, transcription-coupled repair and translesion synthesis. Nucleotide excision repair (NER), which includes both global genome repair and trans-coupled repair, can recognize and excise bulky helix-distorting lesions. Deficiencies in the NER pathway lead to several genetic disorders related to DNA repair, such as XP, trichothiodystrophy (TTD) and Cockayne syndrome (CS) [8].
Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder that is genetically heterogeneous due to seven complementation groups with defective NER and a variant group (XP-V) with normal NER [9]. Xeroderma pigmentosum complementation group E (XP-E) is one of the least common groups of XP. Mutations in the DDB2 gene are responsible for XP-E. DDB2 protein binds to DDB1 to form the UV-DDB complex, which recognizes UV-induced DNA damage and recruits proteins of the global repair pathway to initiate DNA repair [10]. The study of human primary cell lines from patients with XP-E containing DDB2 mutations revealed that DDB2 was proposed to regulate the p53-mediated apoptotic pathway after UV irradiation [11]. XP-E patients usually have a normal sunburn response, but they have an increased frequency of skin cancer, such as basal cell carcinomas, squamous cell carcinoma and melanomas. Sunlight protection from an early age is of significant importance to prevent skin cancer in XP-E patients.
To the best of our knowledge, only 17 XP-E patients have been reported until now [12,13,14]. The mutations of DDB2 identified in all XPE patients are summarized in Table 1. Substitution mutations (eleven of seventeen) were the most frequent mutation type. Mutations occur most frequently in exon 7 (five of eighteen) of DDB2. The frequency of XP-E is higher in white patients. Our patient is the only Chinese patient with XPE. Compared with other patients, our patients had fewer tumours and better prognosis within the one-year follow-up. The reason for the phenotypic variations in XPE patients may depend on climate and lifestyle.
In this study, we identified a novel homozygous mutation, c.111_112del (p.A39Efs*6), of the DDB2 gene in an adult XP-E patient from a Chinese consanguineous family and summarized all mutations of DDB2. This is the first report of XP-E in Chinese people. After an exact diagnosis, strict sun protection was suggested, and the patients received a benefit from this management.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the [https://pan.baidu.com/] repository, https://pan.baidu.com/s/1h6PBB2QV_DGHYLUmF-ztow , access code: tai6. Usename: yrforbmc; password: yangrui417.
Abbreviations
- XP:
-
Xeroderma pigmentosum
- XPE:
-
Xeroderma pigmentosum group E
References
Moriwaki S, Kanda F, Hayashi M, et al. Xeroderma pigmentosum clinical practice guidelines. J Dermatol. 2017;44(10):1087–96.
Kleijer WJ, Laugel V, Berneburg M, et al. Incidence of DNA repair deficiency disorders in western Europe: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst). 2008;7:744–50.
Tamura D, DiGiovanna JJ, Kraemer KH. Founder mutations in xeroderma pigmentosum. J Invest Detmatol. 2010;130:1491–3.
Emmert S. Xeroderma pigmentosum, Cockayne’s syndrome, and trichothiodystrophy. In: Irvine AD, Hoeger P, Yan A, editors. Harper’s Textbook of Pediatric Dermatology. Oxford (UK): Chapt. 135: WileyBlackwell; 2011. p. 135.1–135.24.
Lehmann J, Seebode C, Martens MC, Emmert S. Xeroderma Pigmentosum – facts and perspectives. Anticancer Res. 2018;38(2):1159–64.
Bradford PT, et al. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2011;48(3):168–76.
Sugasawa K. Regulation of damage recognition in mammalian global genomic nucleotide excision repair. Mutat Res. 2010;685:29–37.
Kobaisi F, Fayyad N, Rezvani HR, Fayyad-Kazan M, Sulpice E, Badran B, et al. Signaling Pathways, Chemical and Biological Modulators of Nucleotide Excision Repair: The Faithful Shield against UV Genotoxicity. Oxid Med Cell Longev. 2019;2019:4654206.
Bukowska B, Karwowski BT. Actual state of knowledge in the field of diseases related with defective nucleotide excision repair. Life Sci. 2018;195:6–18.
Wakasugi M, Shimizu M, Morioka H, Linn S, Nikaido O, Matsunaga T. Damaged DNA-binding protein DDB stimulates the excision of cyclobutane pyrimidine dimers in vitro in concert with XPA and replication protein a. J Biol Chem. 2001 May 4;276(18):15434–40.
Itoh T, O’Shea C, Linn S. Impaired regulation of tumor suppressor p53 caused by mutations in the xeroderma pigmentosum DDB2 gene: mutual regulatory interactions between p48 DDB2 and p53. Mol Cell Biol. 2003;23(21):7540–53.
Oh KS, Emmert S, Tamura D, DiGiovanna JJ, Kraemer KH. Multiple skin cancers in adults with mutations in the XP-E (DDB2) DNA repair gene. J Invest Dermatol. 2011;131(3):785–8.
Fassihi H, Sethi M, Fawcett H, et al. Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc Natl Acad Sci U S A. 2016;113(9):E1236–45.
Ben Rekaya M, Naouali C, Messaoud O, et al. Whole exome sequencing allows the identification of two novel groups of Xeroderma pigmentosum in Tunisia, XP-D and XP-E: impact on molecular diagnosis. J Dermatol Sci. 2018;89(2):172–80.
Acknowledgements
We would like to thank the patient and her family for their collaboration.
Funding
This study was supported by the Leading Talents and Innovation Team of Jiangsu Province (CXTDA2017038). The funding body had no influence on study design, data analysis, interpretation of data and writing the manuscript.
Author information
Authors and Affiliations
Contributions
RY performed the experiments and wrote the manuscript. QTK and YYD collected and analysed the clinical data. WWL revised the manuscript. HS designed the study, conducted experiments and reviewed the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Jinling Hospital, and written informed consent was obtained from all participants.
Consent for publication
Written informed consent for publication was obtained from the patients or their guardians.
Competing interests
The authors have no conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, R., Kong, Q., Duan, Y. et al. Identification of a novel DDB2 mutation in a Chinese Han family with Xeroderma pigmentosum group E:a case report and literature review. BMC Med Genet 21, 67 (2020). https://doi.org/10.1186/s12881-020-00997-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-00997-0