
Abstract
Background
Xeroderma pigmentosum (XP) is a rare autosomal recessive skin disorder characterized by hyperpigmentation, premature skin aging, ocular and cutaneous photosensitivity with increased risk of skin tumors. XP is caused by mutations in DNA repair genes that protect cells from UV-induced DNA damage. The current study aims to investigate, on clinical and genetic basis, Moroccan XP patients. We explored by direct sequencing the involvement of the prevalent XPA and XPC genes mutations: nonsense mutation (c.682C>T, p.Arg228X) and a two-base-pair (2 bp) deletion (c.1643 1644delTG or p.Val548Ala fsX25), respectively, in 40 index cases from 37 unrelated families in Moroccan population.
Results
Early skin and ocular manifestations were detected with high rate of malignancy. Cutaneous lesions progressed to malignant skin tumor in 70% of cases. Ocular tumors were also observed in 11 patients including BCC in eight cases, SCC in three cases and melanoma in four cases. Among the 40 patients, there were 20 homozygous cases for the 2 bp deletion in the XPC gene and 9 homozygous cases carrying the nonsense XPA mutation.
Conclusion
These findings obtained in the present study revealed that the XPC gene mutation (c.1643 1644delTG, p.Val548AlafsX25) is the major cause of Xeroderma pigmentosum in our population. The c.682C>T (p.Arg228X) mutation is relatively associated with moderate phenotype in XP group A Moroccan families. This result will also contribute to improving the molecular diagnosis of XP disease and will have a significant impact on improving the care of Moroccan patients and their relatives.
Similar content being viewed by others


Background
Xeroderma pigmentosum (XP; OMIM: 278700–278780) is a rare autosomal recessive skin disorder. XP has been reported around the world with variable incidence. In fact, much higher rates were observed in North African countries (Morocco 1:80,504; Tunisia 1:10,000) compared to US and Western Europe ones (2–3 per million births) [1,2,3,4]. It is mainly due to high frequency of consanguineous marriage in these countries. In fact, the percentage of consanguinity in Morocco reaches 19% with an even greater proportion in the remote regions of the Middle Atlas (25.4%) [5]. Moreover, in XP, the sexes are affected equally
XP was first described by Hungarian dermatologist Moritz Kaposi in 1874. He describes the dry and tightly stretched appearance of the skin of a young German patient. Almost a century after, the relationship between XP symptoms, DNA damage caused by ultraviolet radiations and deficiency of DNA repair mechanism was raised by Gartler in 1964 and Cleaver in 1968. XP is clinically manifested by photosensitivity with an early hyperpigmentation in sun-exposed areas. Furthermore, signs of poikilothermic skin changes and premature skin aging can also be observed at early childhood. The risk of skin cancers, such as BCCs, SCCs and cutaneous melanoma, is particularly increased in patients with XP. These cutaneous manifestations are often associated with ocular lesions and sometimes neurological symptoms [6, 7].
In the past decades, it has been established that XP pathology is mainly caused by mutations in genes encoding for proteins involved in the nucleotide excision repair pathway (NER). The NER is a mechanism for detecting and repairing lesions in the DNA, which are mainly cyclobutane pyrimidine dimers (CPDs) and 6-pyrimidine 4-pyrimidone (6-4PPs) photoproducts. Defective NER fails to excise DNA damage lesions leading to distortion of DNA structure and consequently disruption of DNA replication and transcription. There are two types of NER mechanisms: GGR, for global genome repair, which is a mechanism that repairs damage throughout the genome and TCR, for transcription-coupled repair, which is a repairing mechanism of transcription-blocking lesions. During the GGR-NER, the lesion recognition step is made by the XPC complex. This complex consists of three subunits: XPC, HR23B, and centrin 2 which localize the deformation of the DNA structure caused by the lesion. In TCR-NER, the RNA polymerase II (RNAPII) coupled with two other proteins CSA and CSB recognizes damage at the transcribed DNA strand. After lesion recognition step, both the GGR and the TCR need the same repair factors to remove the lesion. The transcription factor II H (TFIIH) is thus recruited. Thanks to the helicase and ATPase activities of its two XPB and XPD subunits, the complex TFIIH opens the double helix at the damage site. The pre-incision complex is further stabilized by XPA binding to the lesion-containing DNA strand and RPA binding to the undamaged strand. To excise the lesion from the damaged DNA strand, the endonuclease XPG and the complex XPF-ERCC1 are recruited. After excision, XPG recruits PCNA and RFC resynthesis factors. Thus, RPA, XPG, RFC and PCNA factors form a structural base allowing the recruitment of DNA polymerase δ, ε, or κ and the beginning of the resynthesis phase of the repair mechanism. After the gap-filling synthesis, DNA ligase I seals the remaining nick [8, 9].
Mutations in any of these proteins from the NER pathways (GGR or TCR) lead to abnormalities in DNA repair that have been noted in multiple clinical syndromes including Xeroderma pigmentosum. Based on which gene is mutated, XP patients are grouped into seven complementation groups (XP-A to XP-G). An eighth group, the XP variant (XP-V) is caused by mutations in polymerase η gene (Pol H) involved in translesion DNA synthesis [10, 11]. Among these different entities, XP-C is the most prevalent complementation group worldwide, followed by XP-A, especially in North African countries [12]. Nowadays, it becomes common practice to perform molecular analysis of the XP genes in order to determine the exact mutation responsible for the clinical symptoms [13].
The current study aims to investigate, on clinical and genetic basis, 40 Moroccan XP patients. The molecular analyses targeted the mutational status of XPA and XPC genes due to their high prevalence in North African population.
Methods
Subjects
This study was approved by the Medical Genetics Department and Dermatology Department of Hassan II University Hospital in Fez, which drains patients from the northeastern region of Morocco.
We collect a sapmle of forty XP patients with different clinical presentations belonging to 37 unrelated Moroccan families were recruited between July 2017 and January 2022. Informed consent was obtained, and personal data and familial history were noted for each patient.
Genetic screening
Peripheral blood samples (10 ml) were collected in EDTA tubes from all recruited patients. Genomic DNA was extracted using Genomic DNA kit (Invitrogen). The quality and quantity of the DNA were controlled by A260⁄A280 using a Nanodrop spectrophotometer (Nanodrop; Fisher Scientific, Wilmington, DE, USA).
Screening of the most common mutations, c.1643_1644delTG (p.Val548AlafsX25) in exon 9 of XPC and c.682C>T (p.Arg228Ter) in exon 6 of XPA, was performed by polymerase chain reaction (PCR), using the following primers: XPC-9F: 5′CCAGGGTGTCTTATAAAGAGG-3′; XPC-9R:5′-CAAGGCCTTACCTCCAAG-3′; XPA-6Fa:5′-GTGAGGTAAGAAAGTAAGTTTGCCAAG-3′; XPA-6Ra:5′-TCTAGCACTCAGCTCCCATCTCTG-3′; XPA-6Fb: 5′-GTTTCAGTGAAGGTCACCTGGC-3′; XPA-6Rb:5′-GGTTGGTAAATGCTCAGTAAATGTTAGC-3′. PCR was carried out in a final reaction volume of 25 µL containing 100 ng of genomic DNA, 5 U of Taq (Invitrogen), 20 pmol of each primer, 50 mM MgCl2, 10 mM dNTP, and 10 × PCR buffer (Invitrogen). PCR conditions were 95 °C for 10 min; 35 cycles of 95 °C for 30 s, 60 °C (exon9-XPC) or 64 °C (exon6a&b-XPA) for 30 s, and 72 °C for 1 min; and extension at 72 °C for 9 min.
The PCR products were first purified using ExoSAPIT™ (USB Corporation, OH, USA) and then sequenced using BigDye Terminator v3.1 and capillary electrophoresis was performed using the Applied Biosystems 3500Dx Genetic Analyzer. (Applied Biosystems, CA, USA).
The chromatogram was analyzed by the Sequencing Analysis SeqA v.5.4 (Applied Biosystems). The sequences thus obtained underwent bioinformatics analysis using the “Nucleotide Blast” alignment program at http://blast.ncbi.nlm.nih.gov.
Results
Clinical finding
This cohort study involved a series of 40 XP Moroccan patients (20 females and 20 males) from 37 unrelated families. All the clinical data are summarized in Table 1. In fact, the age of disease onset ranged from 0.16 to 22 years with an average of 4.21 years. The age at examination ranged from 1 to 33 years with an average of 9.67 years. Consanguinity was registered in 75% of families (30/40). Among them, 66.67% (20/30) were of the first cousin and 33.33% (10/30) of the second cousin.
All patients developed classic skin manifestations related to XP including skin photosensitivity, poikilotherma and xerosis. In the present study, 24 patients (60%) presented with lentigines localized in photoexposed regions and 62.5% of the cases (25/40) presented with warts. Benign and precancerous skin lesions were also noted in the present study with the presence of keratoacanthoma in 25% of the cases and actinic keratosis in 55%. Furthermore, all patients presented with abnormalities: photophobia in 100%, conjunctivitis in 32.5% and ectropion in 17.5%. Seven patients had also neurological symptoms consisting mainly of neurodevelopmental delay observed in six patients and only one patient presented with a mild dysfunction of the prefrontal cerebral cortex. Benign vascular proliferations, like telangiectasia, were also noted.
Cutaneous lesions progressed to malignant skin tumor in 70% (28/40) of the cases. A total of 245 BCC and 39 SCC were detected in addition to 24 skin melanoma. We diagnosed basal cell carcinoma (BCC) in 25 patients, squamous cell carcinoma (SCC) in 16 patients and melanoma in nine patients. In addition, ocular tumors were also observed in 11 patients including BCC in eight cases, SCC in three cases and melanoma in four cases. The number and distribution of tumors in our cohort are represented in Table 2.
In this cohort, ten patients do not present any XPA or XPC mutation, despite the presence of characteristic clinical manifestations. It is therefore essential to complete the molecular investigation to search for possible mutations characteristic of other XP groups.
Genetic findings
Mutation analysis of XPC gene revealed the presence of the most common deletion of two bases TG in the exon 9 at position c.1643_1644 (NM_004628) in 21/40 cases. 20/40 patients were homozygous for this mutation, whereas one patient presented heterozygous mutation.
The occurrence of hematologic, thyroid and gynecological malignancies has been described in XPC patients in several studies [14,15,16,17]. Nevertheless, none of these clinical manifestations were found among the 21 XPC delTG patients. Rigorous follow-up of these patients and regular blood tests appear essential for a better detection.
Neurological manifestations are noted in 14 to 40% of XP patients, particularly in groups A, B, D and G. There is no correlation between the severity of the skin lesion and the presence of neurological manifestations [18]. However, in our cohort, the XP33 patient had a homozygous mutation del TG along with neurological manifestations.
Furthermore, sequencing analysis showed that 9/40 XP patients presented the recurrent nonsense mutation c.682C>T (p.Arg228Ter) in exon 6 of XPA in homozygote state. However, 5/40 patients were negative for both XPC and XPA mutations.
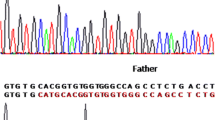
Among the nine patients carrying the c.682C>T mutation of the XPA gene, three present the c.1496C>T (rs2228000) polymorphism in the heterozygous state. Moreover, six patients without mutations present this same polymorphism, two are in the heterozygous state and four in the homozygous state (Table 1, Fig. 1).

Sequencing chromatograms of XPC and XPA mutations identified in our cohort. a wild type, b mutation c.1643-1644delTG at a homozygous state sequence and c mutation c.1643-1644delTG at a heterozygous state, d wild type, e polymorphism C.1496 C>T at a homozygous state sequence and f polymorphism C.1496 C>T at a heterozygous state, g wild type, h mutation C.682C>T at a homozygous state sequence
Discussion
In this study, we described the mutational and clinical profiling of 40 XP cases. It presents, as far as we know, the first large study of XP in the North-East population of Morocco.
The mean age of XP diagnosis was 9.67 years which is consistent with previous studies [7, 19]. However, the mean age for the development of the first skin cancer in our Moroccan cohort was 12.34 years. This average age difference compared to other studies [7, 19] could be explained by the fact that the patients attended at the university hospital present at a relatively advanced age, after a long history of cumulative sun exposure without rigorous sun protection. In fact, six patients presented at an age above 18 years. Globally, our clinical features were quite similar to those reported in several cohorts [9, 19,20,21].
In another side, skin tumors occurred in 70% (28/40) of patients nearly alike other studies [7, 16, 20, 22]. We also noted that all patients who developed skin cancer developed at least one non-melanoma skin cancer (NMSC); either basal or squamous cell carcinoma in sunexposed skin areas. A total of 245 BCC and 39 SCC were detected in addition to 24 skin melanoma. Indeed, few data reported that the frequency of cutaneous tumors differs from one complementation group to the other with a predominance of BCC and SCC in XPC group. They describe the development of higher rate of NMSC compared to melanoma [7]. Thus, our results perfectly correlate with these findings.
This high rate of malignant skin tumors in our Moroccan series in particular and in the Maghreb series in general can be explained by the high cost of strict photoprotection, especially for poor patients, the difficulty in accessing healthcare and the lack of knowledge of the pathology.
Furthermore, the severity of ocular manifestations is generally correlated with that of skin involvement and there is a risk of development of malignant ocular tumors [23, 24]. In our series, 27.5% of patients developed ocular malignancies. Moussaid et al. [25] found that 25.3% of patients presented ocular tumors. North African series reported a rate of approaching 25% [26, 27].
The diagnosis of XP is mainly based on clinical features. Molecular genetics is essential for the determination of the exact complementation group. In North Africa and Southern Europe, XP-C and XP-A are the most frequent groups [21].
The present study focused on two common mutations: the frame shift mutation (c.1643 1644delTG, p.Val548AlafsX25), located in exon 9 of the XPC gene, and the nonsense mutation (c.682C>T) in exon 6 of the XPA gene. The XPC mutation was detected in 21 of 40 (52.5%), and the nonsense mutation in XPA gene was found in 9 of 40 (22.5%) patients.
Molecular investigation of Soufir et al. conducted on 66 unrelated families from the Maghreb region revealed mutations in the XPC gene in 85% of patients; among them, 87% shared the mutation (c.1643 1644delTG). Alterations in the XPA gene were found in 12% of cases with a frequency of about 87.5% for the mutation (c.682C>T) [12]. Senhaji et al. [22] identified XPC mutations in 76% of patients. Bensenouci et al. [20] observed this common mutation in 89.5%. The detected mutation leads to a premature termination codon and the absence of normal XPC protein. Due to the large spread of this mutation in North African countries, several studies put the emphasis on the possibility of having the same ancestor. As a matter of fact, it was shown that this mutation occurred approximately 1250 years ago applying microsatellite haplotyping. This was the time when Muslims from the Arabian Peninsula conquered Southern Europe. Our patients with this mutation also originated from this geographical region [19].
On another side, it was established that patients with typical c.682C>T XP-A mutation exhibit moderate clinical abnormalities and neurological symptoms. The same manifestations were detected in American, European and Japanese families who are sharing this punctual mutation. In fact, this mutation occurs in the C-terminal domain of XPA protein explaining the moderate phenotype of XPA group.
Among the nine patients presenting the XPA common mutation, six showed neurodevelopmental delay. The XP33 patient had a homozygous mutation del TG along with neurological manifestations. The same case was previously described by khan et al. 2009 who described two XPC patients with homozygous mutation in the initiation codon with and without neurological involvement. Possible hypothesis explaining the neurological abnormalities in these patients could be related to the simultaneous inheritance of other pathogenic genes or could result from other gene-modifying effects; rather than the influence of the XPC gene itself [28].
We identified 10 patients with the main clinical manifestations of XP in which none of the previous mutations was detected. Therefore, it is essential to complete the molecular investigation in search of characteristic mutations in the other XP genes.
The interest of molecular diagnosis is based on the early management of patients in the preclinical phase, but also the screening of asymptomatic carriers in order to reduce the frequency of heterozygosity and therefore the incidence of XP through targeted family planning. In this context, all patients and their relatives underwent genetic counseling based on the results of the molecular study.
Conclusion
The results obtained in the present study revealed that the XPC gene mutation (c.1643 1644delTG, p.Val548AlafsX25) is the major cause of Xeroderma pigmentosum in our population. The design of a simple method to allow accurate and early molecular diagnosis in newborns is primordial in order to initiate photoprotection measures before clinical manifestations. Additionally, early diagnosis might improve prognosis of the disease. This could also facilitate genetic counseling for families at risk and facilitate screening of asymptomatic carriers to decrease the frequency of heterozygosity and thus the frequency of Xeroderma pigmentosum.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- XP:
-
Xeroderma pigmentosum
- DNA:
-
Desoxyribonucleic acid
- Arg:
-
Arginine
- Bp:
-
Base pair
- Del:
-
Deletion
- Val:
-
Valine
- Ala:
-
Alanine
- OMIM:
-
Online Mendelian Inheritance in Man
- BCC:
-
Basal cell carcinoma
- SCC:
-
Squamous cell carcinoma
- NER:
-
Nucleotide excision repair
- CPD:
-
Cyclobutane pyrimidine dimer
- 6-4PP:
-
6-Pyrimidine 4-pyrimidone
- GGR:
-
Global genome repair
- TCR:
-
Transcription-coupled repair
- RNAPII:
-
Ribonucleic polymerase II
- CSA:
-
Cockayne syndrome complementation group A
- CSB:
-
Cockayne syndrome complementation group B
- TFIIH:
-
Transcription factor II H
- RPA:
-
Replication protein A
- PCNA:
-
Proliferating cell nuclear antigen
- RFC:
-
Replication factor C
- EDTA:
-
Ethylenediamine tetraacetic acid
- PCR:
-
Polymerase chain reaction
References
Doubaj Y, Laarabi FZ, Elalaoui SC, Barkat A, Sefiani A (2012) Carrier frequency of the recurrent mutation c.1643_1644delTG in the XPC gene and birth prevalence of the Xeroderma Pigmentosum in Morocco. J Dermatol 39(4):382–384
Halim NB, Bouafif NBA, Romdhane L, Atig RKB, Chouchane I, Bouyacoub Y et al (2013) Consanguinity, endogamy, and genetic disorders in Tunisia. J Community Genet 4(2):273–284
Kleijer WJ, Laugel V, Berneburg M, Nardo T, Fawcett H, Gratchev A et al (2008) Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum Cockayne syndrome and trichothiodystrophy. DNA Repair 7(5):744–750
Lehmann J, Schubert S, Emmert S (2014) Xeroderma pigmentosum: diagnostic procedures, interdisciplinary patient care, and novel therapeutic approaches. J Dtsch Dermatol Ges J Ger Soc Dermatol JDDG 12(10):867–872
Romdhane L, Mezzi N, Hamdi Y, El-Kamah G, Barakat A, Abdelhak S (2019) Consanguinity and inbreeding in health and disease in north African populations. Annu Rev Genomics Hum Genet 20(1):155–179
Lucero R, Horowitz D (2022) Xeroderma Pigmentosum. In: StatPearls [Internet]. Treasure Island: StatPearls Publishing [cité 10 mars 2022]. Disponible sur: http://www.ncbi.nlm.nih.gov/books/NBK551563/
Baykal C, Atcı T, Yılmaz Z, Büyükbabani N (2021) Skin tumors in xeroderma pigmentosum: evaluation of a large series and a literature review. J Cutan Pathol 48(7):884–895
Holick MF (2020) Sunlight, UV radiation, vitamin D, and skin cancer: how much sunlight do we need? In: Reichrath J (Eds) Sunlight, vitamin D and skin cancer [Internet]. Cham: Springer International Publishing [cité 7 mars 2022]. pp 19–36. (Advances in Experimental Medicine and Biology). Disponible sur: https://doi.org/10.1007/978-3-030-46227-7_2
Rabie E, Amr K, Zada S, El-Sayed H, El Darouti M, El-Kamah G (2021) Clinical and mutational spectrum of Xeroderma Pigmentosum in Egypt: identification of six novel mutations and implications for ancestral origins. Genes 12(2):295
Schubert S, Lehmann J, Kalfon L, Slor H, Falik-Zaccai TC, Emmert S (2014) Clinical utility gene card for: Xeroderma pigmentosum. Eur J Hum Genet EJHG. https://doi.org/10.1038/ejhg.2013.233
Lehmann J, Seebode C, Martens MC, Emmert S (2018) Xeroderma Pigmentosum–facts and perspectives. Anticancer Res 38(2):1159–1164
Soufir N, Ged C, Bourillon A, Austerlitz F, Chemin C, Stary A et al (2010) A prevalent mutation with founder effect in xeroderma pigmentosum group C from north Africa. J Invest Dermatol 130(6):1537–1542
Lehmann AR, Fassihi H (2020) Molecular analysis directs the prognosis, management and treatment of patients with xeroderma pigmentosum. DNA Repair 93:102907
Oetjen KA, Levoska MA, Tamura D, Ito S, Douglas D, Khan SG et al (2020) Predisposition to hematologic malignancies in patients with xeroderma pigmentosum. Haematologica 105(4):e144–e146
Nikolaev S, Yurchenko AA, Sarasin A (2022) Increased risk of internal tumors in DNA repair-deficient xeroderma pigmentosum patients: analysis of four international cohorts. Orphanet J Rare Dis 17(1):104
Jerbi M, Ben Rekaya M, Naouali C, Jones M, Messaoud O, Tounsi H et al (2016) Clinical, genealogical and molecular investigation of the xeroderma pigmentosum type C complementation group in Tunisia. Br J Dermatol 174(2):439–443
Kouatcheu SD, Marko J, Tamura D, Khan SG, Lee CR, DiGiovanna JJ et al (2021) Thyroid nodules in xeroderma pigmentosum patients: a feature of premature aging. J Endocrinol Invest 44(7):1475–1482
Brooks PJ (2007) The case for 8,5′-cyclopurine-2′-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience 145(4):1407–1417
Schäfer A, Hofmann L, Gratchev A, Laspe P, Schubert S, Schürer A et al (2013) Molecular genetic analysis of 16 XP-C patients from Germany: environmental factors predominately contribute to phenotype variations. Exp Dermatol 22(1):24–29
Bensenouci S, Louhibi L, De Verneuil H, Mahmoudi K, Saidi-Mehtar N (2016) Diagnosis of Xeroderma Pigmentosum groups A and C by detection of two prevalent mutations in west Algerian population: a rapid genotyping tool for the frequent XPC mutation c.1643_1644delTG. BioMed Res Int 2016:2180946
Ben Rekaya M, Jerbi M, Messaoud O, Ben Brick AS, Zghal M, Mbarek C et al (2013) Further evidence of mutational heterogeneity of the XPC gene in Tunisian families: a spectrum of private and ethnic specific mutations. BioMed Res Int 2013:316286
Senhaji MA, Abidi O, Nadifi S, Benchikhi H, Khadir K, Rekaya MB et al (2013) c.1643_1644delTG XPC mutation is more frequent in Moroccan patients with Xeroderma Pigmentosum. Arch Dermatol Res 305(1):53–57
Narang A, Reddy JC, Idrees Z, Injarie AM, Nischal KK (2013) Long-term outcome of bilateral penetrating keratoplasty in a child with xeroderma pigmentosum: case report and literature review. Eye Lond Engl 27(6):775–776
Ramkumar HL, Brooks BP, Cao X, Tamura D, DiGiovanna JJ, Kraemer KH et al (2011) Ophthalmic manifestations and histopathology of Xeroderma Pigmentosum: two clinicopathological cases and a review of the literature. Surv Ophthalmol 56(4):348–361
Moussaid L, Benchikhi H, Boukind EH, Sqalli S, Mouaki N, Kadiri F et al (2004) Tumeurs cutanées au cours du xeroderma pigmentosum au Maroc. Ann Dermatol Vénéréol 131(1):29–33
Khatri ML, Bemghazil M, Shafi M, Machina A (1999) Xeroderma pigmentosum in Libya. Int J Dermatol 38(7):520–524
Bouadjar B, Aït-Belkacem F, Daya-Grosjean L, Sarasin A, Larbaoui SL, Ferhat R et al (1996) Xeroderma pigmentosum. A study in 40 Algerian patients. Ann Dermatol Venereol 123(5):303–306
Khan SG, Oh KS, Emmert S, Imoto K, Tamura D, Digiovanna JJ et al (2009) XPC initiation codon mutation in xeroderma pigmentosum patients with and without neurological symptoms. DNA Repair 8(1):114–125
Acknowledgments
We thank the subjects and their families for participating in this study.
Funding
This work was financially supported by the Hassan II University Hospital, Morocco.
Author information
Authors and Affiliations
Contributions
MA, HS, LB contributed to conception and supervision of study. MA and HS were involved in research techniques. MA and ST contributed to analysis and interpretation of the data. MA and NS were involved in writing of the paper. LB and FZM contributed to critical review. LB, HBB, FZM were involved in clinical assessment. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written consent was obtained from all participants. This study was approved by the local ethic committee located in the Faculty of Medicine and Pharmacy of Fez. The research was conducted in accordance with the principles of the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abbassi, M., Sayel, H., Senhaji, N. et al. Clinical and molecular characterization of Xeroderma pigmentosum in Moroccan population: a case series of 40 patients. Egypt J Med Hum Genet 23, 154 (2022). https://doi.org/10.1186/s43042-022-00368-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00368-9