
Abstract
Background
We sought to analyse MMACHC variants among 126 pedigrees with cobalamin (cbl) C deficiency and combined methylmalonic aciduria and homocystinuria by Sanger sequencing, characterize the spectrum of MMACHC gene variants, and perform prenatal genetic diagnosis by chorionic villus sampling among these pedigrees.
Methods
Peripheral blood was collected from 126 probands and their parents who visited the Genetic Counseling Clinic at our hospital between January 2014 and December 2017, and DNA was extracted from the blood. Then, we amplified the coding sequence and splicing regions of the MMACHC gene by PCR, and the PCR products were further sequenced to detect the variants in each pedigree. In 62 families, pregnant women were subjected to chorionic villus sampling for prenatal genetic diagnosis.
Results
In total, 31 distinct variants were detected in the 126 pedigrees, and the most frequent variants were c.609G > A (p.Trp203Ter), c.658_660delAAG (p.Lys220del), c.567dupT (p.Ile190Tyrfs*13) and c.80A > G (p.Gln27Arg). Two of these variants have not been previously reported in the literature. One variant [c.463_465delGGG (p.Gly155del)] is a small-scale deletion, and the other variant [c.637G>T(p.Glu213Ter)] is a nonsense mutation. Among the 62 pedigrees who received a prenatal diagnosis, 16 foetuses were normal, 34 foetuses were carriers of heterozygous variants, and the remaining 12 foetuses harboured compound heterozygous variants or homozygous variants. Couples whose foetuses were normal or carriers continued the pregnancy, whereas couples whose foetuses harboured compound heterozygous variants or homozygous variants decided to terminate the pregnancy. The follow-up results were consistent with the prenatal diagnosis.
Conclusions
Two novel MMACHC variants were identified, and prenatal genetic diagnosis is an accurate and convenient method that helps avoid the delivery of combined methylmalonic aciduria and homocystinuria patients.
Similar content being viewed by others
Background
Methylmalonic acidaemia or aciduria (MMA), which is the most common inborn error of organic acid metabolism [1], is inherited as an autosomal recessive disease caused by deficiency in methylmalonyl coenzyme A mutase (MCM) or intracellular cobalamin (cbl) metabolism [2]. Based on the patients’ biochemical features, MMA can be classified as isolated MMA or combined methylmalonic aciduria and homocystinuria, and the latter consists of five subtypes, including cblC, cblD, cblF, cblJ and cblX deficiencies [3, 4]. These defects result in methylcobalamin and adenosylcobalamin dysfunction, leading to the subsequent accumulation of methylmalonic acid and homocysteine in the blood and urine, which is a biochemical hallmark of this disorder [5, 6]. In China, the most commonly affected patients have combined methylmalonic acidaemia and homocystinuria, accounting for approximately 80% of all MMA cases. Among the subtypes, the cblC defect (MIM 277400) is the most common [7] and is caused by mutations in the MMACHC gene, which is located in chromosome region 1p34.1; the gene contains five exons and encodes a 282 amino acid protein [8].
An increasing number of cblC patients have benefited from an early diagnosis and better outcomes through expanded newborn screening, particularly gas chromatography-mass spectrometry (GC-MS) [9]. Despite the reduced mortality and morbidity in MMA children, their quality of life remains very low, and the families of these patients suffer a significant economic burden. The most effective measure to reduce the social and family pressure is to avoid the birth of children with MMA by prenatal diagnosis. Measuring the activity of methylmalonyl CoA mutase in amniotic fluid, amniotic fluid cells and chorionic cells and quantifying cbl metabolites in amniotic fluid cells have been employed in prenatal diagnosis [10, 11]. Combined with these biochemical analyses, genetic analyses are also currently used. Other studies report that prenatal diagnosis is performed through simple genetic diagnosis. Here, our study employs a simple genetic diagnosis.
To enhance our knowledge of this disorder, we recruited 126 pedigrees with combined methylmalonic acidaemia and homocystinuria who visited the Genetic Counseling Clinic of the First Affiliated Hospital of Zhengzhou University between January 2014 and December 2017. The MMACHC variants in the probands and their parents were analysed. Among these pedigrees, 62 pregnant mothers underwent prenatal genetic diagnosis. The primary aim of this study was to provide a foundation for rapid and efficient genetic-based diagnosis, genetic counselling for patients with cblC deficiency, and prenatal diagnosis of MMA in China.
This study was approved by the Scientific and Experimental Research Medical Ethics Committee of the First Affiliated Hospital of Zhengzhou University. All analysed samples were obtained after signed informed consent was provided.
Methods
Pedigrees and MMA diagnosis
Our study includes samples from 126 couples who had a child birth history of MMA or children with MMA whose diagnosis was established based on the clinical presentation, positive methylmalonic acid results or newborn screening by GC-MS (GC-MS, QP2010, Schimadzu, Japan). The patients were recruited from the Genetic Counseling Clinic of the First Affiliated Hospital of Zhengzhou University between January 2014 and December 2017. No patients were the product of consanguineous marriages.
DNA extraction
Peripheral blood samples were collected from 90 probands and 126 couples. For families with a deceased-proband, samples were only collected from the parents. Chorionic villus sampling from pregnant women was performed with ultrasonic guidance at 11 to 14 weeks of gestation. Genomic DNA was extracted from the peripheral blood samples and chorionic villus samples using a DNA extraction kit (Omega blood/tissue DNA kit, Georgia, United States) according to the manufacturer’s instructions.
PCR and sanger sequencing
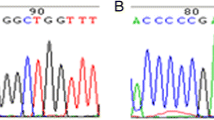
The polymerase chain reaction (PCR) primers were designed according to previously published data [12]. The coding exons and splicing regions of the MMACHC gene were amplified by PCR, and the PCR products were subsequently sequenced bi-directionally using an ABI 3130-xl gene analyser (Life Technologies, Carlsbad, CA, United States). To identify the nucleotides, the sequences were aligned and inspected using a reference sequence from Ensemble (NM_015506) (http://asia.ensembl.org/).
Genotype analysis
By searching the HGMD database (http://www.hgmd.cf.ac.uk/ac/index.php) and SNP database (http://www.ncbi.nlm.nih.gov/), the novel variants were named according to the international gene mutation nomenclature system (http://www.HGVS.org/varnomen). Mutation taster, Polymorphism phenotyping (PolyPhen) and PROVEAN were calculated to predict the pathogenic effects of the variations of interest.
Prenatal diagnosis
After ascertaining the genotype of the probands and their parents, prenatal diagnosis was performed by chorionic villus sampling of pregnant women. To exclude contamination from the mother, a PowerPlex 16 HS System kit (Promega, Madison, WI, USA) was used. The results were analysed using ABI 3130xl and GeneMapper v3.2 software.
Follow-up
Umbilical cord blood was collected for genetic diagnosis of the foetuses.
Results
MMACHC gene variant spectrum
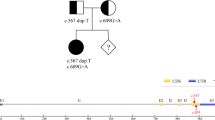
The 4 coding exons of the MMACHC gene and their flanking regions in each sample were analysed. Of the fully genotyped patients, 94 cases harboured compound heterozygous variants, and the remaining 32 cases harboured homozygous variants. All parents were heterozygous carriers.
In total, we identified 31 different variants among the 126 patients recruited for our study. Two of these variants were novel, whereas the remaining 29 variants have been previously described. A complete list of all variants is provided in Table 1. The two allele variants of 126 pedigrees in our study is provided in (Additional file 1: Table S3). Among these variants, the most prevalent variant was c.609G > A(p.Trp203Ter) (48.41%), followed by c.658_660delAAG(p.Lys220del) (13.49%), c.567dupT(p.Ile190Tyrfs*13) (6.75%), c.80A > G (p.Gln27Arg) (5.95%), c.482G > A(p.Arg161Gln) (5.95%) and c.217C > T(p.Arg73Ter) (3.97%). None of the variants described above were identified among 100 healthy control subjects.
The variants can be classified into the following different types: missense variants, nonsense variants, splicing variants, small insertions, and small deletions. The spectrum of these distinct variants was distributed throughout the 4 coding exons and splicing regions of the MMACHC gene. Many variants were located in exon 4 (84.13%), and variants occurring in other exons were less frequent.
Among the 31 different variants, two variants, including the nonsense variant c.637G>T resulting in p.Glu213Ter and the small deletion variant c.463_465delGGG resulting in p.Gly155del, were novel variants. These two variants have not been reported in the HGMD database (http://www.hgmd.org) or the SNP database (http://www.ncbi.nlm.nih.gov/SNP). Moreover, according to the prediction tools PolyPhen and Mutation taster, all novel variants were predicted to be disease causing. Regarding the c.463_465delGGG variant, a conservation analysis of the amino acid sequence of MMACHC from humans and other species, such as P. troglodytes, M. mulatta, F. catus, and M. musculus, revealed that the G155 residue is highly conserved. The deletion of this amino acid may cause the loss of original protein features. The c.637G>T variant is a nonsense variant leading to the translation of a significantly truncated protein. The above-mentioned analysis further confirmed the pathogenicity of these mutations.
Prenatal diagnosis and follow-up
After ascertaining the genotype of these pedigrees, prenatal diagnosis was performed by chorionic villus sampling of pregnant women. Contamination from the mother was prevented using a PowerPlex 16 HS System kit (Promega, Madison, WI, USA).
Among the 62 pedigrees who received prenatal diagnosis, 16 foetuses were normal, 34 foetuses were carriers of heterozygous variants, and the remaining 12 foetuses harboured compound heterozygous variants or homozygous variants (Table 2). Couples whose foetuses were normal or carriers continued the pregnancy, whereas couples whose foetuses harboured compound heterozygous variants or homozygous variants decided to terminate the pregnancy. The gene analysis of the foetuses’ umbilical cord blood was consistent with the prenatal diagnosis. In addition, no abnormal results were noted in newborns screened by GC-MS.
Discussion
This study illustrates the spectrum of variants in the MMACHC gene in 126 pedigrees with cblC defects, including 62 families that choose to undergo prenatal diagnosis by chorionic villus sampling. In total, we identified 31 different variants in these 126 patients. Two of these variants were novel, whereas the remaining 29 variants have been previously reported in the literature.
MMA is the most prevalent organic aciduria involving intermediate metabolism due to enzymatic block. Based on the age at the presentation of the initial symptoms and diagnosis (from newborn to older than 20 years), patients are divided into the following two categories: early-onset and late-onset cblC deficiency. The latter is a rare disorder, and diagnosis is often delayed. Most reported patients with the cblC type present in the first year and typically in the neonatal period with lethargy, vomiting, hypotonia, respiratory distress and developmental delay, which is referred to as early-onset cblC [13, 14]. The other type is the late-onset cblC form, and patients with cblC exhibit ataxia, dementia, psychosis and other neurologic abnormalities generally after 4 years of age [15, 16].
Currently, newborns are screened by MS and GC-MS, and abnormalities in early-onset and late-onset children can be easily observed in urine and blood. Early diagnosis and treatment are beneficial in children with cblC defects, resulting in improved quality of life and reduced suffering from pain.
Treatment options like betaine, hydroxocobalamin, methylfolate, vitamin B6 and L-carnitine are applied to these patients clinically, the combined treatment can effectively improve the clinical manifestations in acute phase and alleviate biochemical abnormalities in hematuria. The median levels of serum homocysteine, and urine methylmalonic acid were significantly decreased (p < 0.01), from 97.3 μmol/L (ranged from 25.1 to 250 μmol/L) and 168.55 (ranged from 3.66 to 1032.82) before treatment to 43.8 μmol/L (ranged from 17 to 97.8 μmol/L) and 6.81 (ranged from 0 to 95.43) after treatment, respectively [17]. For those from a rural area they suffer more pain due to undertreatment, or late initiation of optimal treatment in China. Even with enough combined treatment, along with much lower levels of serum homocysteine and urine methylmalonic acid than before, their long-term outcomes are not encouraging, along with severe neurological sequelae in most patients.
To date, more than 90 mutations in this gene, including missense mutations, nonsense mutations, small indels, and splicing region mutations, have been reported in the literature [18]. Given the high allelic heterogeneity in MMA, the spectrum of variants differs in various populations worldwide. Thus, mutational analysis of MMA patients is crucial in different populations. Specifically, c.271dupA (21%), c.394C > T (21%) and c.609G > A (11%) are the most frequent variants in Europe based on a study conducted in 2014 [19]. Other studies have reported that the c.394C > T variant is the most common allele in Native American and Middle Eastern MMA patients [20, 21]. However, numerous children with the cblC type have been diagnosed as homozygous for the c.609G > A variant, and more importantly, these children were East Asians [12, 20]. Another Chinese study reported that 55.4% (51/92) of all variants were c.609G > A [22]. Similarly, in this study, c.609G > A accounted for 48.41% of the variants. Among the 32 homozygous variants, 29 variants were homozygous for c.609G > A. Thus, we further confirm that c.609G > A is a very common mutation in China. Following the most common c.609G > A variant is c.658_660delAAG, which accounted for 13.49% in this study and is also a very common mutation in Chinese patients [8].
Of the 31 different variants identified in this study, 2 variants were novel. One variant, i.e., the nonsense variant c.637G>T (p.Glu213Ter), results in a truncated protein at residue 213. The other variant, i.e., small deletion c.463_465delGGG (p.Gly155del), causes the deletion of a glycine at residue 155. The c.463G > C (Gly155Arg) and c.464G > A (Gly155Glu) variants have been previously reported to be pathogenic [23]. In addition, the highly conserved results of the PROVEAN and PolyPhen analyses of glycine 155 support the pathogenicity of the deletion. However, further research should be performed to verify our hypothesis.
Although early detection and treatment can improve the outcome in cblC patients, most probands die at a young age. Thus, preimplantation genetic diagnosis or screening (PGD/PGS) or prenatal diagnosis are the best currently available prevention methods. MMA can be diagnosed as early as the first trimester by chronic villus sampling, which is beneficial for families with probands.
However, undeniably, this study also has some limitations in terms of prenatal diagnosis. First, in families with deceased probands, we first detected the MMACHC gene variants in the parents. If both parents harboured a heterozygous variant of the MMACHC gene, we deduced that the proband genotype harboured compound heterozygous or homozygous variants. Direct testing of the probands was not performed. Second, traditional methods used for prenatal diagnosis include biochemical detections, such as measuring the activity of methylmalonyl CoA mutase in the amniotic fluid, amniotic fluid cells and chorionic cells and quantifying cbl metabolites in amniotic fluid cells. Our study exclusively performed a simple genetic diagnosis to these pedigrees with explicit homozygous or compound heterozygous mutations without traditional biochemical measurements. However, for those pedigrees with only one or no definite deleterious alleles, or some variants of unknown significance, we suggest them to other Hospital to perform prenatal diagnosis with biochemical testing, so here we do not collect them into our study, and we are unable to help them. Finally, pregnant women bearing MMA foetuses typically choose to terminate the pregnancy, which also represents a type of injury to the pregnant women. Overall, preimplantation genetic diagnosis can be useful for avoiding miscarriages or the induction of labour in early pregnancy and is a future direction in the development of MMA prenatal diagnosis.
Conclusion
Two novel variants of the MMACHC gene were identified, and prenatal genetic diagnosis is an accurate and convenient method to help avoid the delivery of combined methylmalonic aciduria and homocystinuria patients.
Abbreviations
- cblC:
-
Cobalamin(cbl) C
- GC-MS:
-
Gas chromatography-mass spectrometry
- MCM:
-
Methylmalonyl coenzyme A mutase
- MMA:
-
Methylmalonic acidemia or aciduria
- PCR:
-
Polymerase chain reaction
- PGD/PGS:
-
Preimplantation genetic diagnosis or screening
References
Chen M, Zhuang J, Yang J, Wang D, Yang Q. Atypical hemolytic uremic syndrome induced by CblC subtype of methylmalonic academia: a case report and literature review. Medicine. 2017;96:e8284.
Fowler B, Leonard JV, Baumgartner MR. Causes of and diagnostic approach to methylmalonic acidurias. J Inherit Metab Dis. 2008;31:350–60.
Zong Y, Liu N, Zhao Z, Kong X. Prenatal diagnosis using genetic sequencing and identification of a novel mutation in MMACHC. BMC Med Genet. 2015;16:48.
Yu HC, Sloan JL, Scharer G, et al. An X-linked cobalamin disorder caused by mutations in transcriptional coregulator HCFC1. Am J Hum Genet. 2013;93:506–14.
Carrillo-Carrasco N, Chandler RJ, Venditti CP. Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management. J Inherit Metab Dis. 2012;35:91–102.
Carrillo-Carrasco N, Venditti CP. Combined methylmalonic acidemia and homocystinuria, cblC type. II. Complications, pathophysiology, and outcomes. J Inherit Metab Dis. 2012;35:103–14.
Liu YP, Ma YY, Wu TF, et al. Abnormal findings during newborn period of 160 patients with early-onset methylmalonic aciduria. Zhonghua er ke za zhi. 2012;50:410–4.
Wang F, Han LS, Hu YH, et al. Analysis of gene mutations in Chinese patients with methylmalonic acidemia and homocysteinemia. Zhonghua er ke za zhi. 2009;47:189–93.
Scolamiero E, Cozzolino C, Albano L, et al. Targeted metabolomics in the expanded newborn screening for inborn errors of metabolism. Mol BioSyst. 2015;11:1525–35.
Zhang Y, Yang YL, Hasegawa Y, et al. Prenatal diagnosis of methylmalonic aciduria by analysis of organic acids and total homocysteine in amniotic fluid. Chin Med J. 2008;121:216–9.
Inoue Y, Ohse M. Prenatal diagnosis of methylmalonic aciduria by measuring methylmalonic acid in dried amniotic fluid on filter paper using gas chromatography-mass spectrometry. Anal Bioanal Chem. 2011;400:1953–8.
Lerner-Ellis JP, Tirone JC, Pawelek PD, et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat Genet. 2006;38:93–100.
Weisfeld-Adams JD, Bender HA, Miley-Akerstedt A, et al. Neurologic and neurodevelopmental phenotypes in young children with early-treated combined methylmalonic acidemia and homocystinuria, cobalamin C type. Mol Genet Metab. 2013;110:241–7.
Yu YF, Li F, Ma HW. Relationship of genotypes with clinical phenotypes and outcomes in children with cobalamin C type combined methylmalonic aciduria and homocystinuria. Zhongguo dang dai er ke za zhi. 2015;17:769–74.
Han B, Cao Z, Tian L, et al. Clinical presentation, gene analysis and outcomes in young patients with early-treated combined methylmalonic acidemia and homocysteinemia (cblC type) in Shandong province, China. Brain Dev. 2016;38:491–7.
Matos IV, Castejon E, Meavilla S, et al. Clinical and biochemical outcome after hydroxocobalamin dose escalation in a series of patients with cobalamin C deficiency. Mol Genet Metab. 2013;109:360–5.
Huang Z, Han LS, Ye J, et al. Outcomes of patients with combined methylmalonic acidemia and homocystinuria after treatment. Zhonghua er ke za zhi. 2013;51:194–8.
Chang JT, Chen YY, Liu TT, Liu MY, Chiu PC. Combined methylmalonic aciduria and homocystinuria cblC type of a Taiwanese infant with c.609G>a and C.567dupT mutations in the MMACHC gene. Pediatrics Neonatol. 2011;52:223–6.
Huemer M, Scholl-Burgi S, Hadaya K, et al. Three new cases of late-onset cblC defect and review of the literature illustrating when to consider inborn errors of metabolism beyond infancy. Orphanet J Rare Dis. 2014;9:161.
Lerner-Ellis JP, Anastasio N, Liu J, et al. Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype-phenotype correlations. Hum Mutat. 2009;30:1072–81.
Nogueira C, Aiello C, Cerone R, et al. Spectrum of MMACHC mutations in Italian and Portuguese patients with combined methylmalonic aciduria and homocystinuria, cblC type. Mol Genet Metab. 2008;93:475–80.
Wang F, Han L, Yang Y, et al. Clinical, biochemical, and molecular analysis of combined methylmalonic acidemia and hyperhomocysteinemia (cblC type) in China. J Inherit Metab Dis. 2010;33(Suppl 3):S435–42.
Komhoff M, Roofthooft MT, Westra D, et al. Combined pulmonary hypertension and renal thrombotic microangiopathy in cobalamin C deficiency. Pediatrics. 2013;132:e540–4.
Funding
This work was supported by grants from internal funding from the First Affiliated Hospital of Zhengzhou University.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files. For the raw data of “The two allele variants of 126 pedigrees in our study”, it is presented in the Additional file 1: Table S3. The data in Tables 1 and 2 were uploaded as a single Item named “Table” in Attach Files section. They could be found in Page 6 and 7 within this published article.
Author information
Authors and Affiliations
Contributions
XK conceived the study. SM summarized all the data and was involved in the histological analysis. NL and XK provided expertise for the data interpretation and suggestions for the manuscript preparation. SH and XK wrote the manuscript. All authors have read and approved the manuscript, and that is the case.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Scientific and Experimental Research Medical Ethics Committee of the First Affiliated Hospital of Zhengzhou University approved this study. All analysed samples were obtained after signed informed consent was provided. For all the minor probands, their parents consent for their participation in this study on their behalf. For the adult patients, all of them consent for their participation.
Consent for publication
All analysed samples were obtained after signed informed consent was provided.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S3. The two allele variants of 126 pedigrees in our study. (DOCX 23 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hu, S., Mei, S., Liu, N. et al. Molecular genetic characterization of cblC defects in 126 pedigrees and prenatal genetic diagnosis of pedigrees with combined methylmalonic aciduria and homocystinuria. BMC Med Genet 19, 154 (2018). https://doi.org/10.1186/s12881-018-0666-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0666-x