
Abstract
Background
Mutations in TSC1 or TSC2 cause the tuberous sclerosis complex (TSC), while mutations in PKD1 or PKD2 cause autosomal dominant polycystic kidney disease (ADPKD). PKD1 lays immediately adjacent to TSC2 and deletions involving both genes, the PKD1/TSC2 contiguous gene syndrome (CGS), are characterized by severe ADPKD, plus TSC. mTOR inhibitors have proven effective in reducing angiomyolipoma (AML) in TSC and total kidney volume in ADPKD but without a positive effect on renal function.
Methods and results
We describe a patient with independent truncating PKD1 and TSC2 mutations who has the expected phenotype for both diseases independently instead of the severe one described in PKD1/TSC2-CGS. Treatment with mTOR inhibitors reduced the AML and kidney volume for 2 years but thereafter they resumed growth; no positive effect on renal function was seen throughout. This is the first case addressing the response to mTOR treatment when independent truncating mutations in PKD1 and TSC2 are present.
Conclusions
This case reveals that although PKD1 and TSC2 are adjacent genes and there is likely cross-talk between the PKD1 and TSC2 signalling pathways regulating mTOR, having independent TSC2 and PKD1 mutations can give rise to a milder kidney phenotype than is typical in PKD1/TSC2-CGS cases. A short-term beneficial effect of mTOR inhibition on AML and total kidney volume was not reflected in improved renal function.
Similar content being viewed by others

Background
Autosomal dominant polycystic kidney disease (ADPKD) is the most common kidney disorder with a Mendelian inheritance pattern, and a prevalence ranging from 1/400 to 1/1000 worldwide [1, 2]. It is responsible for 4–10 % of end-stage renal disease (ESRD) in Western countries [3, 4]. ADPKD shows both locus and allelic heterogeneity. Two causative genes—PKD1, and PKD2 have been identified [5, 6]. The TSC2 and PKD1 genes are overlapped at their 3′ UTR ends by 3 bp. The TSC2 gene encodes tuberin and together with TSC1, encoding for hamartin, causes the tuberous sclerosis complex (TSC). TSC is an autosomal dominant disorder, with high penetrance and a birth incidence of 1 in 6000–11,000 [7]. Clinically, it manifests with skin lesions, renal, neurological, pulmonary and cardiac symptoms. For the adequate diagnosis the established diagnostic criteria by Northrup et al. should be followed [8]. The clinical presentation of TSC ranges from a few features of the disease to severe neurological involvement [9].
Analysis of TSC2 patients with severe renal cystic disease showed they can have deletions also disrupting PKD1; a contiguous gene syndrome (CGS). Brook-Carter et al. identified 6 TSC2 children with very severe polycystic disease showing deletions that involved both genes [10].
These children, as well as others reported in the literature, present with enlarged polycystic kidneys recognizable in utero, at birth or shortly thereafter [10–12]. Their kidneys are filled by a multitude of variably sized cysts, closely resembling those seen in advanced stages of ADPKD and they usually enter ESRD in the second or third decade of life. Other typical lesions of TSC, such as angiomyolipomas (AMLs), ungual fibromas, and well-established facial angiofibromas, only appear later in life [11–17]. No description of patients having both diseases caused by two independent mutations in the PKD1 and the TSC2 genes have been reported to date.
Tuberin and hamartin form a complex that regulates signaling through the mammalian target of rapamycin (Rheb/mTOR/p70S6K) pathway, which controls processes such as cell growth, cell cycle progression and apoptosis. Mutations to TSC1 or TSC2 permit aberrant upregulation of mTOR signaling causing increased protein synthesis and cell growth [18]. Also, polycystin 1 (PC1), the PKD1 protein product, interacts and protects tuberin S939 from AKT phosphorylation and helps to retain tuberin in the membrane to suppress mTOR activity [19–21].
Inhibition of mTOR has been proposed as therapeutic approach for both TSC and ADPKD. To date, the results are promising for TSC but are not encouraging for ADPKD [22–28].
We report here a patient with TSC and ADPKD due to independent mutations in both TSC2 and PKD1 who was treated with mTOR inhibitors showing a good response based on AML and cystic burden decrease but without preservation of renal function. The cross talk between tuberin, hamartin, the polycystins and mTOR are discussed to explain the phenotype of the patient and his response to mTOR inhibition.
Methods
A 26-year-old man first presented to our renal unit at 11 years of age following detection of cystic kidneys.
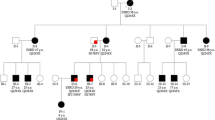
His father, paternal aunt, paternal grandmother and sister have ADPKD (Fig. 1). The age at onset of ESRD was 68 for the grandmother, 44 for the father and 48 for the aunt. The patient’s sister has normal renal function, hypertension and enlarged kidneys (kidney length 17.5 cm) at the age of 30. There is no family history of TSC.

Panel a Pedigree of the family showing the segregation analysis of haplotypes as well as PKD1 and TSC2 mutations. The arrow points the proband reported in this case Panel b MLPA for PKD1 gene and the 3′ end of TSC2 gene, each bar represents the normalized peak height for the probe indicated on the x axis. The heavy black lines represent the deletion of the PKD1 exons 1–10 in heterozygosis. Panel c AML volume evolution: 1A and 1B baseline; 2A and 2B at the end of 3 years treatment with mTOR inhibitors; 3A and 3B one year later (without treatment). The AML decreased in size after 3 years on treatment and slightly increased in size one year after treatment withdrawal. Panel d Right (top row) and left kidney (bottom row): initial MR (1R and 1L), after 3 years on treatment with mTOR inhibitors (2R and 2L) and 1 year later (without treatment) (3R and 3L)
The patient was diagnosed with TSC at 3 months due to hypomelanic macules and a seizure. An echocardiogram revealed a cardiac rhabdomyoma, which was removed at 6 months. A brain MRI showed numerous subependymal nodules and periventricular calcifications. A retinal astrocytoma was also detected in the left eye and abnormal retinal vessels in the right one. Facial angiofibroma developed in early childhood. Development progressed normally with no further seizures or mental retardation. A kidneys ultrasound scan performed at 3 years demonstrated multiple small cysts throughout the renal parenchyma. Serial yearly ultrasound scans showed an AML of 3 cm of diameter in the left kidney at the age of 14. Cyst size and number increased along with the AML, which was 6 cm with a kidney length of 17 cm at 22 years. Because of concerns about the increasing size of the AML, local ethical approval was obtained and sirolimus started at 22 years of age (mean dose: 3 mg/day, trough levels 6.9 ± 3.8 ng/ml).
The patient and his family signed informed consents allowing researchers to publish their data and imaging. They also signed informed consent for the genetic study. The study was approved by the IRB of Fundació Puigvert.
Imaging
Abdominal imaging evaluations were performed by 1.5 Tesla magnetic resonance (Vantage Atlas, Toshiba Medical Systems Corporation, Otawara-shi, Tochigi-ken, JAPAN) with a body phased-array coil. All studies were performed with the patient in supine position. Coronal, sagittal and axial scans were acquired with T1-weighted fast spoiled gradient echo and T2-weighted fast spin echo protocols with and without fat suppression.
Abdominal studies were analyzed by two independent radiologists with more than 10 years of experience interpreting abdominal imaging studies. Before the start of the evaluation, the radiologists showed an intra and inter-observer variability of less than 5 % in focal renal mass measurement.
The volumes of individual kidneys were measured in T1-weighted images with use of the stereologic method [25, 29, 30]. Tumour volume was estimated using a standardised validated software program (Vitrea, Vital Imaging version 4.1.14.0).
Genetic studies
Haplotype analysis was performed using microsatellite markers within and closely flanking PKD1/TSC2 and PKD2. The markers used were: HBAP1, D16S3024, D16S3395, KG8, AC2.5, CW2, D16S3070 for PKD1/TSC2 and D4S1534, D4S2462, D4S2929, D4S2460, D4S423 for PKD2. The analyses of these markers were performed by PCR amplification using fluorescent primers and resolved on the ABI 3130-Avant Genetic Analyzer.
Mutation screening of PKD1 and TSC2 genes were performed by direct Sanger sequencing. The duplicated region of PKD1 was amplified as five PKD1-specific fragments by long-range polymerase chain reaction (LR-PCR) followed by nested PCRs [31] combined with Sanger sequencing of all 46 PKD1 exons. For TSC2, the 42 exons were amplified and sequenced with primers designed using genomic sequence information (GenBank accession number: NG_005895.1) and the Primer 3 (v. 0.4.0) program [32]. To screen for PKD1/PKD2 deletion/duplication Salsa MLPA kit P351-B1/P352-B1 (MRC-Holland, Amsterdam, Netherlands) was used, which also includes probes for exons 35, 37 and 41 of the TSC2 gene.
Results
Six months after commencing treatment, there was a reduction in the volume of the AMLs and renal volume on MRI scan, which continued decreasing for 18 more months, although increased during the third year (Table 1) (Fig. 1c). The number of facial angiofibromas remained unchanged, but the lesions were smaller, paler and less rough. At that time, the negative results of two large trials using mTOR inhibitors in ADPKD were released, which together with the evidence of regrowth under treatment, prompted us to discontinue the mTOR inhibition.
One year after discontinuation, the AML had further increased along with the renal volume, and the GFR decreased (Table 1). The only side effect of the drug was an increase of the protein to creatinine ratio during treatment which decreased after cessation (Table 1). Although normal blood pressure was recorded while on treatment, when it stopped a slight but abnormal increase in diastolic BP was detected and treatment with ACEI was started achieving a good control of BP.
Initially linkage analysis showed that the ADPKD family was linked to the PKD1 gene (Fig. 1). Also, heterozygosity of the KG8 microsatellite marker (located in the 3′ region of PKD1) ruled out a contiguous gene syndrome. Subsequently, sequencing and MLPA analysis of the PKD1 and TSC2 genes, disclosed a deletion of PKD1 (exons 1–10, Fig. 1b): c.1-?_2097 + ?del, (p.Met1fs) (definitely pathogenic mutation), and a TSC2 nonsense mutation: TSC2 c.2251C > T (p.Arg751*) (definitely pathogenic mutation). The PKD1 mutation was present in all affected members of the family while the TSC2 mutation was absent in all but the proband.
Discussion
A single individual carrying mutations in two different genes causing two different diseases, and therefore suffering both entities, is not frequent but depends on the prevalence of each disease. Taking into account that the prevalences of ADPKD and TSC are around one in 800 and one in 8.000, respectively, the probability for the simultaneous occurrence of these two diseases is approximately one in 6.400.000 births. Due to difficulty of genetic testing to demonstrate the coexistence of mutations in the two genes, probably this unusual population has been under represented in the literature.
Most patients with TSC and severe cystic disease have a CGS with a deletion involving the coding regions of both the TSC2 and the PKD1 genes (PKD1/TSC2-CGS) [10, 14, 33, 34]. They consist of ~3 % of TSC patients overall [9]. The prevalence of kidney cysts in TSC alone is between 30 and 50 %, more frequent in TSC2 than TSC1 [9, 26, 35], especially when the TSC2 gene harbours a nonsense or frameshift mutation [36]. But these non-CGS TSC patients generally have just a few small cysts of little clinical relevance [17, 37] and, do not present renal failure [37]. However, children with PKD1/TSC2-CGS usually enter ESRD in the second or third decade of life [10, 17]. Nevertheless, some PKD1/TSC2-CGS patients have milder renal disease [13, 38] with mosaicism being the main reason which is rather common in these cases but not supported by the blood derived DNA studies here [12, 17, 37].
A case with separate TSC1 and PKD2 mutations had a very modest cystic phenotype, probably due to the relatively mild disease in PKD2 compared to PKD1 and because TSC1 mutations are rarely associated with renal cysts [39].
The case we present here has a de novo nonsense mutation in the TSC2 gene and a germ line deletion of the first 10 exons of the PKD1 gene. The TSC2 mutation appeared spontaneously in this patient and does not show mosaicism in blood but we cannot rule out mosaicism at the organ level, particularly in the kidney. ADPKD in this family is quite severe but what is most surprising is the apparent limited impact of an additional TSC2 mutation in the proband. He will probably enter ESRD sooner than his relatives but not by many years. In fact, his kidney size does not differ significantly from his sister. If the CGS phenotype were just due to an additive effect of disrupting both genes, one would expect the same effect of a truncating mutation in both genes than a deletion involving both of them. However, as some cases of late onset of ESRD have been described in PKD1/TSC2-CGS the real explanation for a milder phenotype remains elusive [5, 13, 38]. Given that the primary transcripts from these genes slightly overlap at the 3′ region, regulation due to antisense noncoding (nc)RNAs or miRNA binding could be unusually altered. One limitation of the present case is the impossibility to know whether the mutations are in cis or in trans due to the fact that the TSC2 mutation is de novo. If the TSC2 mutation is in cis position with the PKD1 deletion, the proband is expected to have similar disease presentation as patients with TSC2/PKD1 CGS. However, if the two mutations are in trans, the second hit, likely by loss of heterozygosity, can only lead to loss of both TSC2 and PKD1 function on one chromosome while leaving one normal copy of both. Under this scenario, the proband will maintain normal function of one of the two genes with a milder phenotype than expected for TSC2/PKD1 CGS. However, as stated by Brook Carter et al., the phenotypes associated with independent deletion of each of the contiguous genes are different and the contiguous syndromes represent an accumulation of these phenotypes [10].
Tuberin, hamartin and PC1 are located at the basal body or primary cilia and have been proposed to form a complex which inhibits mTOR [20]. Alternatively, the CGS phenotype may be due to disruption of two independent cystogenic mechanisms, PC1 involving cilia and tuberin through proliferation. Hartman found that Rapamycin enhanced cilia formation in TSC1 and TSC2 null cells and concluded that the efficacy of mTOR inhibitors on renal cystic disease in patients carrying a TSC mutation or PKD1/TSC2-CGS may differ from its efficacy in ADPKD [40]. The fact that some cysts in ADPKD tissue and Pkd1 mutant kidneys do not appear to upregulate mTORC1 and the small number of cysts in patients with TSC calls into question the essential role of the mTOR cascade in cyst formation [41]. Trials have demonstrated efficacy of mTOR inhibitors in TSC with clear reduction in the volume of AML [23–26]. However, mTOR inhibitor studies in ADPKD patients have not been successful [27, 28]. In AML, reduction of volume and probably vascularisation is an excellent end point, but for ADPKD a positive impact on renal function as well as a reduction in kidney volume is ideally required. This is the first described case treated with mTOR inhibitors having TSC and ADPKD and showed that mTOR inhibition reduced AML volume and cystic volume during the first 2 years, but renal function still declined. Interestingly, both AML and renal volume increased during the third year even while on the treatment with adequate Rapamycin plasma levels, which may be due to an adaptive escape mechanism from mTOR inhibition, although this is an uncommon event in non-cancer cells. This patient experienced an increase in the protein/creatinine ratio which may be explained by the effect of mTOR inhibition. The exact mechanism by which mTOR inhibitors affect glomerular permeability is not known. Many mechanisms have been propose such as, decreased VEGF synthesis and expression, dose-related alteration of podocyte slit diaphragm-associated protein structure and activation of the innate immune system, resulting in an increased number of glomerular macrophages [42–47].
Longer term trials in PKD1/TSC2-CGS patients would be interesting in light of our results and also it would be interesting to test the levels of mTOR activation in the peripheral blood of classical CGS patients and of this specific case.
Conclusions
Although PKD1 and TSC2 are adjacent genes and tuberin-hamartin and PC1 may cross-talk and regulate mTOR inhibition, having independent mutations in TSC2 and PKD1 does not necessarily give rise to the typically severe PKD1/TSC2-CGS phenotype. However this is a unique case and the mild disease presentation needs further verification when additional cases become available.
mTOR inhibitors are efficient in reducing AML and ADPKD kidney volume, but do not have a positive impact on renal function.
References
Iglesias CG, Torres VE, Offord KP, Holley KE, Beard CM, Kurland LT. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935–1980. Am J Kidney Dis. 1983;2:630–9.
Dalgaard OZ. Bilateral polycystic disease of the kidneys; a follow-up of two hundred and eighty-four patients and their families. Acta Med Scand Suppl. 1957;328:1–255.
Collins AJ, Foley RN, Herzog C, Chavers B, Gilbertson D, Ishani A, et al. US Renal Data System 2010 Annual Data Report. Am J Kidney Dis. 2011. doi:10.1053/j.ajkd.2010.10.007.
Catalan Transplant Organisation HD GdC. Catalan Renal Registry. 2011.
The European Polycystic Kidney Disease Consortium. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994;78.
Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–42.
Osborne JP, Fryer A, Webb D. Epidemiology of tuberous sclerosis. Ann N Y Acad Sci. 1991. doi:10.1111/j.1749-6632.1991.tb37754.x.
Northrup H, Krueger DA. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243–54.
Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006. doi:10.1056/NEJMra055323.
Brook-Carter PT, Peral B, Ward CJ, Thompson P, Hughes J, Maheshwar MM, et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease–a contiguous gene syndrome. Nat Genet. 1994;8:328–32.
Torra R, Badenas C, Darnell A, Camacho JA, Aspinwall R, Harris PC, et al. Facilitated diagnosis of the contiguous gene syndrome: tuberous sclerosis and polycystic kidneys by means of haplotype studies. Am J Kidney Dis. 1998;31:1038–43.
Consugar MB, Wong WC, Lundquist PA, Rossetti S, Kubly VJ, Walker DL, et al. Characterization of large rearrangements in autosomal dominant polycystic kidney disease and the PKD1/TSC2 contiguous gene syndrome. Kidney Int. 2008;74:1468–79.
Smulders YM, Eussen BH, Verhoef SWC. Large deletion causing the TSC2-PKD1 contiguous gene syndrome without infantil polycytic disease. J Med Genet. 2003;40:2–4.
Martignoni G, Bonetti F, Pea M, Tardanico R, Brunelli M, Eble JN. Renal disease in adults with TSC2/PKD1 contiguous gene syndrome. Am J Surg Pathol. 2002;26:198–205.
Mancino C, Balducci A. Tuberous sclerosis complex and early-onset autosomal dominant polycystic kidney disease as a “contiguous gene” syndrome: report of a case. Contrib Nephrol. 1997;122:96–7.
Culty T, Molinie V, Lebret T, Savareux L, Souid M, Delahousse M, et al. TSC2/PKD1 contiguous gene syndrome in an adult. Minerva Urol Nefrol. 2006;58:351–4.
Sampson JR, Maheshwar MM, Aspinwall R, Thompson P, Cheadle JP, Ravine D, et al. Renal cystic disease in tuberous sclerosis: role of the polycystic kidney disease 1 gene. Am J Hum Genet. 1997;61:843–51.
Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–94.
Distefano G, Boca M, Rowe I, Wodarczyk C, Ma L, Piontek KB, et al. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol Cell Biol. 2009;29:2359–71.
Weimbs T. Regulation of mTOR by polycystin-1: is polycystic kidney disease a case of futile repair? Cell Cycle. 2006;5:2425–9.
Dere R, Wilson PD, Sandford RN, Walker CL. Carboxy terminal tail of polycystin-1 regulates localization of TSC2 to repress mTOR. PLoS One. 2010;5:e9239.
Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008. doi:10.1056/NEJMoa063564.
Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:817–24.
Davies DM, de Vries PJ, Johnson SR, McCartney DL, Cox JA, Serra AL, et al. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: a phase 2 trial. Clin Cancer Res. 2011;17:4071–81.
Cabrera-Lopez C, Marti T, Catala V, Torres F, Mateu S, Ballarin J, et al. Assessing the effectiveness of rapamycin on angiomyolipoma in tuberous sclerosis: a two years trial. Orphanet J Rare Dis. 2012;7:87.
Dabora SL, Franz DN, Ashwal S, Sagalowsky A, DiMario Jr FJ, Miles D, et al. Multicenter phase 2 trial of sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors regress VEGF-D levels descrease. PLoS One. 2011;6, e23379.
Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:820–9.
Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:830–40.
Chapman AB, Guay-Woodford LM, Grantham JJ, Torres VE, Bae KT, Baumgarten DA, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int. 2003. doi:10.1046/j.1523-1755.2003.00185.x.
Bae KT, Commean PK, Lee J. Volumetric measurement of renal cysts and parenchyma using MRI: phantoms and patients with polycystic kidney disease. J Comput Assist Tomogr. 2000;24:614–9.
Rossetti S, Chauveau D, Walker D, Saggar-Malik A, Winearls CG, Torres VE, et al. A complete mutation screen of the ADPKD genes by DHPLC. Kidney Int. 2002;61:1588–99.
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115.
Oyazato Y, Iijima K, Emi M, Sekine T, Kamei K, Takanashi J, et al. Molecular analysis of TSC2/PKD1 contiguous gene deletion syndrome. Kobe J Med Sci. 2011;57:E1–10.
Longa L, Brusco A, Carbonara C, Polidoro S, Scolari F, Valzorio B, et al. A tuberous sclerosis patient with a large TSC2 and PKD1 gene deletion shows extrarenal signs of autosomal dominant polycystic kidney disease. Contrib Nephrol. 1997;122:91–5.
Yates JRW. Tuberous sclerosis. Eur J Hum Genet. 2006. doi:10.1038/sj.ejhg.5201625.
Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C, Maat-Kievit A, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet. 2005;13:731–41.
Kozlowski P, Roberts P, Dabora S, Franz D, Bissler J, Northrup H, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007. doi:10.1007/s00439-006-0308-9.
Boehm D, Bacher J, Neumann HP. Gross genomic rearrangement involving the TSC2-PKD1 contiguous deletion syndrome: characterization of the deletion event by quantitative polymerase chain reaction deletion assay. Am J Kidney Dis. 2007;49:e11–21.
Woerner AC, Au KS, Williams AT, Harris PCNH. Tuberous sclerosis complex and polycystic kidney disease together: an exception to the contiguous gene syndrome. Genet Med. 2006;8:197–8.
Hartman TR, Liu D, Zilfou JT, Robb V, Morrison T, Watnick T, et al. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet. 2009;18:151–63.
Yoder BK, Tousson A, Millican L, Wu JH, Bugg Jr CE, Schafer JA, et al. Polaris, a protein disrupted in orpk mutant mice, is required for assembly of renal cilium. Am J Physiol Ren Physiol. 2002;282:F541–52.
Schonenberger E, Ehrich JH, Haller H, Schiffer M. The podocyte as a direct target of immunosuppressive agents. Nephrol Dial Transplant. 2011;26:18–24.
Cina DP, Onay T, Paltoo A, Li C, Maezawa Y, De Arteaga J, et al. Inhibition of MTOR disrupts autophagic flux in podocytes. J Am Soc Nephrol. 2012;23:412–20.
Letavernier E, Legendre C. mToR inhibitors-induced proteinuria: mechanisms, significance, and management. Transplant Rev. 2008;22:125–30.
Kirsch AH, Riegelbauer V, Tagwerker A, Rudnicki M, Rosenkranz AR, Eller K. The mTOR-inhibitor rapamycin mediates proteinuria in nephrotoxic serum nephritis by activating the innate immune response. Am J Physiol Renal Physiol. 2012;303:F569–75.
Diekmann F, Andres A, Oppenheimer F. mTOR inhibitor-associated proteinuria in kidney transplant recipients. Transplant Rev. 2012;26:27–9.
Stallone G, Infante B, Pontrelli P, Gigante M, Montemurno E, Loverre A, et al. Sirolimus and proteinuria in renal transplant patients: evidence for a dose-dependent effect on slit diaphragm-associated proteins. Transplantation. 2011;91:997–1004.
Acknowledgements
The present study has been funded by: ISCIII: RETIC REDinREN/RD06/0016 and RD012/0021 FEDER funds, FIS PI12/01523, FIS PI13/01731) and Novartis Pharmaceutical.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors had no involvements that might raise the question of bias in the work reported or in the conclusions, implications, or opinions stated.
Authors’ contributions
CC did most of the patient’s follow up. GB and EA did the genetic studies, conceived the study and drafted the manuscript. TM and VC did the MRI interpretations. RT conceived of the study, and participated in its design and coordination and helped to draft the manuscript. JB, JJB and PCH helped interpreting results. All authors contributed to elaborating the manuscript. All authors read and approved the final manuscript.
Authors’ information
RT chairs the Inherited Renal Diseases Unit at Fundació Puigvert. She also chairs the working group on inherited kidney disorders within the Spanish Society of Nephrology and is the President of the Scientific Committee for the AIRG-E.
Elisabet Ars and Roser Torra contributed equally to this work.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Cabrera-López, C., Bullich, G., Martí, T. et al. Insight into response to mTOR inhibition when PKD1 and TSC2 are mutated. BMC Med Genet 16, 39 (2015). https://doi.org/10.1186/s12881-015-0185-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-015-0185-y