
Abstract
Background
Plasmodium vivax apical membrane antigen-1 (pvama-1) is an important vaccine candidate against Malaria. The genetic composition assessment of pvama-1 from wide-range geography is vital to plan the antigen based vaccine designing against Malaria.
Methods
The blood samples were collected from 84 P. vivax positive malaria patients from different districts of Khyber Pakhtunkhwa (KP) province of Pakistan. The highly polymorphic and immunogenic domain-I (DI) region of pvama-1 was PCR amplified and DNA sequenced. The QC based sequences raw data filtration was done using DNASTAR package. The downstream population genetic analyses were performed using MEGA4, DnaSP, Arlequin v3.5 and Network.5 resources.
Results
The analyses unveiled total 57 haplotypes of pvama-1 (DI) in KP samples with majorly prevalent H-14 and H-5 haplotypes. Pairwise comparative population genetics analyses identified limited to moderate genetic distinctions among the samples collected from different districts of KP, Pakistan. In context of worldwide available data, the KP samples depicted major genetic differentiation against the Korean samples with Fst = 0.40915 (P-value = 0.0001), while least distinction was observed against Indian and Iranian samples. The statistically significant negative values of Fu and Li’s D* and F* tests indicate the evidence of population expansion and directional positive selection signature. The slow LD decay across the nucleotide distance in KP isolates indicates low nucleotide diversity. In context of reference pvama-1 sequence, the KP samples were identified to have 09 novel non-synonymous single nucleotide polymorphisms (nsSNPs), including several trimorphic and tetramorphic substitutions. Few of these nsSNPs are mapped within the B-cell predicted epitopic motifs of the pvama-1, and possibly modulate the immune response mechanism.
Conclusion
Low genetic differentiation was observed across the pvama-1 DI among the P. vivax isolates acquired from widespread regions of KP province of Pakistan. The information may implicate in future vaccine designing strategies based on antigenic features of pvama-1.
Similar content being viewed by others


Background
Malaria is an acute febrile infectious disease caused by vector-borne apicomplexan parasites of the genus Plasmodium. The P. vivax and P. falciparum are predominant species responsible for malaria [1]. The P. vivax is most widely distributed human malaria parasite, endemic in tropical and subtropical countries of Asia, South Pacific, Central and South America, Middle East, and North Africa [2]. According to the latest WHO report, about 229 million cases and 40,900 deaths occur due to malaria in 2019 [3].
Treatment and control of malaria have become a serious challenge due to drug resistance and lack of effective vaccines. The wide-range distribution, antigenic variation, relapsing and co-infection led to a collective interest towards the development of effective vaccine against P. vivax [4]. The implementation of RTS,s/AS01 vaccination was started in three African countries during 2019 and considered effective against malaria to date. Furthermore, the R21/Matrixs-M vaccine was tested among children in Bukina Faso and reported to meet the WHO’s goal up to 77% against malaria [5]. Several antigens of Plasmodium species such as apical membrane antigen-1 (AMA-1), Circumsporozoite proteins (CSP), Merozoite surface proteins (MSP) and Duffy binding protein (DBP) are reported as potent malarial vaccine candidates’ targets [6].
The genetic composition assessment of vaccine candidates’ loci is indispensable in modern-age to plan an effective vaccination strategy. There are ample of studies suggesting the AMA-1 of Plasmodium species as promising malaria vaccine candidate antigens [7]. The AMA-1 is a type-I integral membrane protein with molecular size of 83 kDa, mainly expressed in the merozoite and sporozoite stages of Plasmodium parasites [8, 9]. The main biological function of AMA-1 is not well known so far, however the stage-specific expression and localization suggest its crucial role during invasion of erythrocytes and hepatocytes by malarial parasites [10,11,12]. The protein consists of cysteine rich ectodomain having three separated domains (i.e. Domain I, II, and III), a conserved cytoplasmic region and a transmembrane domain [13]. The ectodomain of the protein is highly immunogenic and evokes natural immune responses among patients infected by P. falciparum and P. vivax [14,15,16,17]. Furthermore, the protein AMA-1 reported to elicit the antibody production that effectively halt the invasion of erythrocytes by malarial parasite and confers protective immune responses [18]. This suggesting the AMA-1 as a leading malarial vaccine candidate.
The domain-I of AMA-1 exhibits high level genetic polymorphism and this region appears to be a major target of anti-AMA-1 protective antibodies [19,20,21,22]. It is therefore noteworthy to monitor genetic variations of the vaccine candidate antigens among global malaria pathogenic isolates circulating in endemic areas, in order to design effective vaccine [23]. Several studies about antigenic variation of Plasmodium vivax ama-1 (pvama-1) have been conducted in malaria endemic countries [24,25,26,27,28]. However, limited studies are reported about pvama-1 genetic features from Pakistan. Particularly, no study till date is reported from remote malaria endemic regions of Khyber Pakhtunkhwa (KP) province of Pakistan. The current study was therefore designed to evaluate the genetic composition of pvama-1 among P. vivax isolates collected from widespread KP regions of Pakistan (Fig. 1).

Map of Khyber Pakhtunkhwa (KP), Pakistan. The different districts from where the samples are collected have been marked
Methods
Study design, samples collection and DNA purification
The current study was approved from ethical review committee of Abdul Wali Khan University Mardan (AWKUM/Biochem/Dept/Commit/ECR/18). Blood samples were obtained from 100 consented patients tested positive for P. vivax using microscopy and rapid diagnostic test, while examined in different hospitals and private sector laboratories from Mardan, Swat, Buner, Hangu, Swabi, Kohat, Bannu, Timergara and Peshawar districts of KP province, Pakistan (Fig. 1). The region have an average annual rain fall of 384 mm during the two malaria seasons from March to May and from August to November. The mean temperature in the region ranges from 20 to 40 °C. The blood samples from the patients were collected prior to treatment, spotted on filters, air-dried, and kept in individual sealed plastic bags at ambient temperature until use. The genomic DNA was extracted from the spotted blood samples using a QIAmp blood kit (Qiagen, CA, USA) according to manufacturer’s instructions. The DNA samples were stored at − 20 °C.
PCR amplification and DNA sequencing of pvama-1 DI
A DNA fragment flanking the DI region of pvama-1 was amplified by polymerase chain reaction (PCR) using the specific primers and amplification conditions as reported previously [21, 29]. The resulted PCR products were analyzed on 1.5% agarose gel, purified, and cloned into the T&A vector (Real Biotech Corporation, Banqiao City, Taiwan). Ligation mixture was transformed into Escherichia coli DH5α competent cells, and positive clones were selected by colony PCR. The nucleotide sequence of cloned insert was analyzed by automatic DNA sequencing with M13 forward and reverse primers (Genotech Inc., Daejoen, Korea). The raw data was filtered for quality assessment using DNASTAR Lasergene package.
DNA polymorphism analyses
The DNA sequences data generated in the current study was analyzed in comparison with reference pvama-1 sequence i.e. Sal-I (AF063138) and Genbank-deposited pvama-1 sequences from China Myanmar Boarder (KX495505–KX495577), Iran (KF422636.1–KF422681.1), Korea (KM230319.1–KM230384.1), Myanmar (FJ157248.1–FJ157285.1), Papua New Guinea (PNG) (KC702402.1–KC702501.1), Sri Lanka (EF218679.1–EF218701.1), Venezuela (EU346015.1–EU346087.1), Thailand (FJ784891.1–FJ784990.1), and India (EU282774.1–EF025196.1). The comparative sequences analyses were performed using MEGA4 software suite [30] to identify and evaluate the polymorphic loci.
Functional prediction of nsSNPs
The BepiPred-2.0 [31] server was used for prediction of Linear B-cell epitopes of pvama-1 with a threshold score of ≥ 0.5. The higher BepiPred score predicts higher binding affinity of epitopes with immune receptors. The non-synonymous SNPs (nsSNPs) mapping within the top predicted epitopes of pvama-1 was checked. The intrinsically unstructured regions (IURs) and RBC binding sites within the pvama-1 have previously been characterized [25, 32]; and their annotation features were adopted in current study to check the nsSNPs mapping within these motifs of pvama-1. Additionally, the positive selection sites in B-cell epitopes were identified via the maximum likelihood method of Codeml [33] implemented in EasyCodeML [34].
Statistical and population genetic analyses
The DnaSP v6.12 software package [35] was used to estimate parsimony informative sites, total number of mutations, pairwise nucleotide diversity (π), segregating sites (S), haplotypes (H) composition of the sequences, haplotypes diversity (Hd), recombination (R) between adjacent nucleotides per generation and the minimum number of recombination events (Rm). Additionally the linkage disequilibrium (LD) was estimated between the various polymorphic sites based on the R2 index via DnaSP [35]. The Tajima’s D, Fu and Li’s D* and F* indices were calculated via a sliding window method using DnaSP. The population genetics statistical analyses, including pairwise fixation index (Fst), analysis of molecular variance (AMOVA), haplotype frequencies, and nucleotide diversity based on Nei’s net distance (DA) were computed using Arlequin v3.5 [36]. The haplotype networking analysis and plot were generated using PopArt software [37].
Results
Genetic polymorphic features of KP pvama-1
The 416 bp sequences of pvama-1, flanking the DI domain were amplified from genomic DNA of 84 P. vivax positive samples. The sequences data spanning the 322–737 nucleotide positions of the reference pvama-1 sequence i.e. Sal-I (AF063138). The analyses identified a large numbers of single nucleotide polymorphisms (SNPs) in KP samples. Among these, the 68 were nsSNPs, i.e. causing amino acid substitutions, including 53 dimorphic, 10 trimorphic, 3 tetramorphic, and 2 pentamorphic nsSNPs. The two pentamorphic amino acid changes observed were R112K/T/E/S and S228D/N/R/K. The ten trimorphic amino acid substitutions include the N132D/G, A141E/G, E145A/G, K190E/Q, T191K/P, A199T/V, S209G/C, P210S/L, P223L/S, and V233L/P. While the three tetramorphic amino acid changes are K120R/S/G, E189N/K/G, and E227V/K/G. These amino acid substitutions were observed at varied frequencies in the KP samples. Among the 68 nsSNPS, 59 have previously been reported in literature for P. vivax isolates from different geographical origin. However, the rest of 9 nsSNPs were found specific to KP samples set of this study. These nsSNPs were observed at low frequencies, i.e. 1.1 to 1.19%. Few nsSNPs, including K120R, N132D, L140I, A141E, K190E, E227V, and S228D were commonly observed in KP samples, as well as in some other continental pvama-1 sequences with high frequency of 3.8–100% (Table 1). The KP pvama-1 DI showed overall haplotype diversity (Hd) of 0.978 ± 0.008 (Additional file 1: Fig. S1). A total of 62 segregating sites (S) and 67 mutations were identified for the samples. The Fu and Li’s D* and F* test and Tajima’s D were calculated to check the deviation from neutrality, and to identify whether natural selection have shaped the genetic composition of the pvama-1 D1 region. The Fu and Li’s D* and F* tests results were found significantly negative for KP samples data (Table 2). The Tajima’s D value was negative, i.e. − 1.490, however not significant (P > 0.10). The difference between dN/dS ratio for pvama-1 DI region was also found negative (− 0.05413 ± 0.02) in case of KP samples set.
Haplotype networking analysis
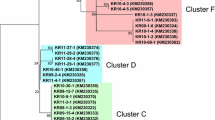
Total 57 haplotypes were identified for the 84 KP samples sequences of pvama-1 DI (Additional file 2: Table S1). The H-14 haplotype was identified with high frequency and shared among samples collected from six different KP districts including, Kohat, Hungo, Buner, Swat, Timergara and Bannu. The H-5 haplotype was identified as second predominant haplotype shared among samples collected from five different KP districts (i.e. Mardan, Swat, Hungo, Bannu and Kohat). The H-3 haplotype was also identified with highest frequency among samples collected from Swat, Mardan, Peshawar and Bannu districts. The pairwise AMOVA inferred genetic distances among haplotypes. The H-53, i.e. predominant in Peshawar samples, was identified as distinct and showed significant genetic differentiation against the H-6 and H-55. The H-6 and H-55 were identified with high frequency in samples collected from Mardan and Peshawar regions respectively. The majorly shared haplotypes of KP samples collected from different districts appeared on share nodes of network plot, however, some haplotypes from Timergara, Peshawar, Kohat and Hungo samples occupied distinct nodes in the network plot, inferring their distinctive features (Fig. 2). The size of each node in haplotype network plot indicates the frequency of a particular haplotype. The length of the line between nodes is proportion to the number of nucleotide substitutions composing the haplotypes.

The Network plot of KP, Pakistan samples based on pvama-1 DI sequences. The circle represents each haplotype, and the size of each circle is proportional to the number of samples holding that specific haplotype. The lines connecting the haplotypes reflect the distance of relatedness between haplotypes. The colors indicate the samples groups collected from different districts of KP, Pakistan
Functional impact assessment of nsSNPs
The functional impact of the nsSNPs (Table 1) was assessed with respect to amino acids substitution in the unstructured/disordered regions, i.e. IURs, RBC binding region and B-cell epitopes of pvama-1. The IURs regions are considering important in vaccine designing and diagnosis. The analysis unveiled that two SNPs i.e., M171T, V172T mapped within the IURs motifs of pvama-1 DI domain. Four SNPs i.e. R240C, N241D, D242E and W243R are mapped within the RBC binding sites of pvama-1. Likewise, 40 nsSNPs, including four novel nsSNPs, i.e. found in newly sequenced KP samples, were mapped within the top predicted B-cell epitopes of pvama-1 (Table 1). The B-cell epitopes were additionally examined for positive selection using the four site-specific models implemented in Easy CodeML [33], i.e. M0—one-ration vs. M3—discrete, M1a—nearly neutral vs. M2a—positive selection, M7 (β) vs. M8—β & ω > 1, and M8a—β & ω = 1 vs. M8—β & ω = 1. The likelihood ratio test (LRTs) was implemented to identify the site-specific positive selection with > 99% posterior probability. Total 15 residues were predicted underlined positive selection with P ≤ 0.05 in the pvama-1 D1 domain region. These include the R112K, K120R, N130K, A131A, N132D, L140L, A141E, E145A, E189E, K190K, H193Y, P210P, E227E, S228S, and V233V. Among these, the nine SNPs, i.e. the R112K, K120R, N130K, A131A, N132D, E145A, P210P, E227E, and S228S are mapped within the B-cell epitopes.
Recombination and linkage disequilibrium (LD) analyses
The KP samples sequences along with global sequences showed decline of LD index R2 with the increase of nucleotide distance which speculate high meiotic recombination events across the pvama-1 region. The R value for KP samples were observed higher compare to China-Myanmar boarder, Korean, and Sri Lankan samples, while lower than the Myanmar samples sequences previously deposited in Genbank [21, 22, 24,25,26, 38,39,40,41]. The higher R value for isolates sequences from different regions depicts the opportunity of high multiclonal infections, cross fertilization and recombination [32]. The higher values of recombination and rapid LD decay as observed in case of some geographical samples speculating the recombination as a possible factor to provoke genetic diversity (Table 3). However, in case of KP samples slow LD decay across the nucleotide distance in pvama-1 DI region was observed that inferring low nucleotide diversity (Additional file 3: Fig. S2).
Nucleotide diversity across pvama-1 in context of global isolates
The sequences of KP isolates (n = 84) were compared to the global pvama-1 sequences deposited in Genbank. The values of K and π observed for KP sequences were more or less similar to previously reported sequences from Iran and India, however different from the rest of global sequences (Table 2). The fixation index Fst statistic was used to assess the genetic differentiation across pvama-1 DI region among KP samples collected from different districts as well as in context of global samples. The pairwise analysis inferred genetic distinction of samples collected from Swabi district compare to rest of the KP regions. The top Fst differentiation was detected between the Bannu and Swabi isolates (Fst = 0.16258, P-value = 0.00977), followed by Swabi and Kohat (Fst = 0.12932; P-value = 0.04199) samples. The lowest Fst was estimated between Swat and Bannu samples (Fst = − 0.07427, P-value = 0.96973), followed by Swat and Hungo samples (Fst= − 0.06635, P-value = 0.89551) (Fig. 3A). The negative Fst is consider zero, depicting no genetic distinction between the population groups. The highest pairwise net number of nucleotide variation (DA) and mean pairwise differences (πxy) was observed between Bannu and Swabi samples, i.e. congruent to the Fst result (Fig. 3B). In context of global samples, marked genetic distinction inferred for KP samples compare to India, Iran, Thailand, Sri-Lanka, Korea, Venezuela, Myanmar, PNG, and China-Myanmar. Highest pairwise genetic differentiation was observed between KP and Korean samples (Fst = 0.40915). The Korean samples showed significant genetic distinction in pairwise comparison to rest of the global samples as well. Meanwhile, least genetic differentiation was observed among KP, Iranian, and Indian samples (Fig. 3C). The highest within population genetic differentiation (π) was found for Korean samples followed by South East Asian samples (Fig. 3D). Pearson correlation plot showed relationship among KP, Sri Lanka, Iran, India and Myanmar samples, congruent to pairwise Fst (Additional file 4: Fig. S3). The plot showed correlation among the populations in hierarchical order. However, the Korean samples showed high genetic distinction in term of Fst value, probably due to geographical separation (Additional file 5: Fig. S4).

Pairwise population genetics analyses. A The graph represents the average number of pairwise differences (πxy), between the KP, Pakistan P. vivax samples population groups collected from different districts (Green above diagonal); within-population group πxx (orange diagonal) and the net number of nucleotide differences between population’s groups (Nei distance DA) (blue below diagonal) based on pvama-1 DI gene variants. B The graph represents the average number of pairwise differences (πxy) between KP, Pakistani samples in context of global samples (Green above diagonal); within-population group πxx (orange diagonal) and the net number of nucleotide differences between population’s groups (Nei distance DA) (blue below diagonal) based on pvama-1 DI variants. C Heat-map plot of pairwise Fst based on pvama-1 gene sequences of KP, Pakistan samples groups collected from different districts. D Heat map of pairwise Fst distinction between KP, Pakistan P. vivax samples groups based on pvama-1 gene sequences in context of worldwide sequences deposited in Genbank, NCBI.
AMOVA test determines the degree of regional variations and homogeneity within and among the isolates. We employed the AMOVA test to determination the genetic variation at single and multiple loci due to variation within a population group as well as between population groups. The AMOVA analysis depicted that genetic diversity in KP samples mainly arose due to within group differentiation i.e. 100.24%, instead of among groups genetic differentiation (− 0.24%). Likewise, higher variance component was noticed within the population group i.e. 3.67534 as compare to among populations (Table 4). Overall, the AMOVA test results inferring less genetic variation among KP isolates, collected from different wide-spread districts of KP region of Pakistan.
Discussion
The comprehensive knowledge about the antigenic variants in Plasmodium parasites is perquisite to design effective vaccine strategies workable in different endemic regions [42]. The current study aimed to analyze genetic composition of pvama-1, a leading malaria vaccine candidate antigen, among P. vivax isolates collected from different districts of KP, Pakistan.
The southern and northern regions of KP province of Pakistan are distinct with respect to geographical and environmental perspectives. However, limited genetic diversity of P. vivax across pvama-1 DI domain was identified in the current study, suggesting no significant genetic heterogeneity among the P. vivax isolates from southern to northern KP regions. The low genetic diversity across the DI domain of pvama-1 in KP regions might arose due to low endemicity of Plasmodium genotypes, as the low endemic region is generally characterized with limited parasitic genetic diversity [43, 44]. The low transmission and endemicity of P. vivax in the KP, Pakistan might have been provoked due to active malaria control programs in these regions from last several years. The pairwise genetic analyses unveiled more or less homogeneous genetic composition of KP samples to South/Central Asian samples from India and Iran regions. This might be due to close geographical contacts among these countries. The excess of segregating sites and significant negative Fu and Li’s D* and F* tests results indicate an excess of rare alleles in KP samples that might result from selective sweep. Additionally these results indicate excess of singleton mutations emerging from rapid population growth and directional selection. More or less similar data pattern was observed for Myanmar isolates [21]. The directional selection lead towards fixing beneficial alleles in the population and causes reduce genetic diversity [45]. However, the negative value (− 0.05413 ± 0.02) of dN/dS ratio for KP isolates data predicted the scenario of purifying and negative selection [37, 38].
The analyses of KP samples in context of global samples inferred unique genetic features and 9 KP samples-specific nsSNPs were identified in the newly sequenced samples. The genetic polymorphisms identified in the current study were further analyzed with respect to their possible functional consequences in the predicted RBC binding sites, B-cell epitopes, and IURs regions of pvama-1. Several nsSNPs were found to be located at the predicted RBC-binding sites, B-cell epitopes and IURs region of pvama-1. The nsSNPs mapped at the B-cell predicted epitopic motifs indicating a high degree of positive selection across the B-cell epitopes region of pvama-1. Likewise, several pvama-1 SNPs were detected in pvama-1 IURs region. The IURs play an important role in molecular recognition, assembling and protein modification [46]. The pvama-1 IURs are indispensable for attachment and invasion of the parasite into RBCs [47]. The protein structure affected by amino acid changes due to these nsSNPs may affect the physicochemical perspectives of the P. vivax AMA-1 protein that might help the parasite to escape from host protective immunity.
Conclusion
The partial DNA sequencing and analyses of pvama-1 DI domain unveiled limited genetic diversity of pvama-1 across the KP regions. This somehow suggested that pvama-1 based vaccine against P. vivax might be promising to effectively combat and contribute in malaria eradication throughout the KP province of Pakistan.
Availability of data and materials
All the relevant data is provided in Additional figures and tables. The entire DNA sequencing data generated during current study is available from Genbank, NCBI (https://www.ncbi.nlm.nih.gov/genbank/) under the accession numbers; ON238020–ON238103.
Abbreviations
- Pv:
-
Plasmodium vivax
- SNPs:
-
Single nucleotide polymorphism
- AMA:
-
Apical membrane antigens
- RBC:
-
Red Blood Cells
- DI:
-
Domain I
- PCA:
-
Principal component analysis
- LD:
-
Linkage disequilibrium
- PCR:
-
Polymerase chain reaction
- KP:
-
Khyber Pakhtunkhwa
References
McFadden GI. Plasmodium: more don’ts. Trends Parasitol. 2019;35(1):4–6.
Mendis K, Sina BJ, Marchesini P, Carter R. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg. 2001;64(1):97–106.
WHO. World malaria report. Geneva: WHO; 2020.
Herrera S, Corradin G, Arévalo-Herrera M. An update on the search for a Plasmodium vivax vaccine. Trends Parasitol. 2007;23(3):122–8.
Datoo MS, Natama MH, Somé A, Traoré O, Rouamba T, et al. Efficacy of a low-dose candidate malaria vaccine, R21 in adjuvant Matrix-M, with seasonal administration to children in Burkina Faso: a randomized controlled trial. Lancet. 2021;397(10287):1809–18.
Dobaño C, Sedegah M, Rogers WO, Kumar S, Zheng H, Hoffman SL, et al. Plasmodium: mammalian codon optimization of malaria plasmid DNA vaccines enhances antibody responses but not T cell responses nor protective immunity. Exp Parasitol. 2009;122(2):112–23.
Thera MA, Coulibaly D, Kone AK, Guindo AB, Traore K, Sall AH, et al. Phase 1 randomized controlled trial to evaluate the safety and immunogenicity of recombinant Pichia pastoris-expressed Plasmodium falciparum apical membrane antigen 1 (PfAMA1-FVO [25–545] in healthy Malian adults in Bandiagara. Malar J. 2016;15(1):1–11.
Healer J, Murphy V, Hodder AN, Masciantonio R, Gemmill AW, Anders RF, et al. Allelic polymorphisms in apical membrane antigen-1 are responsible for evasion of antibody‐mediated inhibition in Plasmodium falciparum. Mol Microbiol. 2004;52(1):159–68.
Silvie O, Franetich JF, Charrin S, Mueller MS, Siau A, Bodescot M, et al. A role for apical membrane antigen 1 during invasion of hepatocytes by Plasmodium falciparum sporozoites. J Biol Chem. 2004;279(10):9490–6.
Peterson MG, Marshall VM, Smythe JA, Crewther PE, Lew AN, Silva AN, et al. Integral membrane protein located in the apical complex of Plasmodium falciparum. Mol Cell Biol. 1989;9(7):3151–4.
Mitchell GH, Thomas AW, Margos G, Dluzewski AR, Bannister LH, et al. Apical membrane antigen 1, a major malaria vaccine candidate, mediates the close attachment of invasive merozoites to host red blood cells. Infect Immun. 2004;72(1):154–8.
Yap A, Azevedo MF, Gilson PR, Weiss GE, O’Neill MT, Wilson DW, et al. Conditional expression of apical membrane antigen 1 in Plasmodium falciparum shows it is required for erythrocyte invasion by merozoites. Cell Microbiol. 2014;16(5):642–56.
Hodder AN, Crewther PE, Anders RF. Specificity of the protective antibody response to apical membrane antigen 1. Infect Immun. 2001;69(5):3286–94.
Udhayakumar V, Kariuki S, Kolczack M, Girma M, Roberts JM, Oloo AJ, et al. Longitudinal study of natural immune responses to the Plasmodium falciparum apical membrane antigen (AMA-1) in a holoendemic region of malaria in western Kenya: Asembo Bay Cohort Project VIII. Am J Trop Med Hyg. 2001;65(2):100–7.
Cortés A, Mellombo M, Masciantonio R, Murphy VJ, Reeder JC, Anders RF. Allele specificity of naturally acquired antibody responses against Plasmodium falciparum apical membrane antigen 1. Infect Immun. 2005;73(1):422–30.
Rodrigues MH, Rodrigues KM, Oliveira TR, Cômodo AN, Rodrigues MM, et al. Antibody response of naturally infected individuals to recombinant Plasmodium vivax apical membrane antigen-1. Int J Parasitol. 2005;35(2):185–92.
Moncunill G, Aponte JJ, Nhabomba AJ, Dobano C. Performance of multiplex commercial kits to quantify cytokine and chemokine responses in culture supernatants from Plasmodium falciparum stimulations. PLoS ONE. 2013;8(1):e52587.
Gentil EC, Damgaard A, Hauschild M, Finnveden G, Eriksson O, Thorneloe S, et al. Models for waste life cycle assessment: review of technical assumptions. Waste Manag. 2010;30(12):2636–48.
Escalante AA, Grebert HM, Chaiyaroj SC, Magris M, Biswas S, Nahlen BL, et al. Polymorphism in the gene encoding the apical membrane antigen-1 (AMA-1) of Plasmodium falciparum. X. Asembo Bay Cohort Project. Mol Biochem Parasitol. 2001;113(2):279–87.
Polley SD, Conway DJ. Strong diversifying selection on domains of the Plasmodium falciparum apical membrane antigen 1 gene. Genet. 2001;158(4):1505–12.
Moon SU, Na BK, Kang JM, Kim JY, Cho SH, Park YK, et al. Genetic polymorphism and effect of natural selection at domain I of apical membrane antigen-1 (AMA-1) in Plasmodium vivax isolates from Myanmar. Acta Trop. 2010;114(2):71–5.
Arnott A, Mueller I, Ramsland PA, Siba PM, Reeder JC, Barry AE, et al. Global population structure of the genes encoding the malaria vaccine candidate, Plasmodium vivax apical membrane antigen 1 (Pv AMA1). PLoS Negl Trop Dis. 2013;7(10):e2506.
Volkman SK, Neafsey DE, Schaffner SF, Park DJ, Wirth DF. Harnessing genomics and genome biology to understand malaria biology. Nat Rev Genet. 2012;13(5):315–28.
Gunasekera AM, Wickramarachchi T, Neafsey DE, Ganguli I, Perera L, Premaratne PH, et al. Genetic diversity and selection at the Plasmodium vivax apical membrane antigen-1 (PvAMA-1) locus in a Sri Lankan population. Mol Biol Evol. 2007;24(4):939–47.
Zhu X, Zhao P, Wang S, Liu F, Liu J, Wang J, et al. Analysis of Pvama1 genes from China-Myanmar border reveal little regional genetic differentiation of Plasmodium vivax populations. Parasites Vectors. 2016;9(1):1–2.
Kang JM, Lee J, Cho PY, Moon SU, Ju HL, et al. Population genetic structure and natural selection of apical membrane antigen-1 in Plasmodium vivax Korean isolates. Malar J. 2015;14(1):1–2.
Heidari A, Keshavarz H, Hajjaran H, Ebrahimi SM, Kabir K, Naseri MH, et al. Genetic variation and selection of domain I of the Plasmodium vivax apical membrane antigen-1 (AMA-1) gene in clinical isolates from Iran. Iran J Parasitol. 2013;8(4):536.
Rajesh V, Elamaran M, Vidya S, Gowrishankar M, Kochar D, Das A. Plasmodium vivax: genetic diversity of the apical membrane antigen-1 (AMA-1) in isolates from India. Exp Parasitol. 2007;116(3):252–6.
Dias S, Somarathna M, Manamperi A, Escalante AA, Gunasekera AM, Udagama PV, et al. Evaluation of the genetic diversity of domain II of Plasmodium vivax apical membrane antigen 1 (PvAMA-1) and the ensuing strain-specific immune responses in patients from Sri Lanka. Vaccine. 2011;29(43):7491–504.
Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24(8):1596–9.
Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45(W1):W24–9.
Kang JM, Lee J, Moe M, Jun H, Lê HG, Kim TI, Thái TL, Sohn WM, Myint MK, Lin K, Shin HJ, Kim TS, Na BK. Population genetic structure and natural selection of Plasmodium falciparum apical membrane antigen-1 in Myanmar isolates. Malar J. 2018;17(1):71.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91.
Gao F, Chen C, Arab DA, Du Z, He Y, Ho SY. EasyCodeML: a visual tool for analysis of selection using CodeML. Ecol Evol. 2019;9(7):3891–8.
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34(12):3299–302.
Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform. 2005;1:117693430500100000.
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48.
Rastaghi AR, Nedaei F, Nahrevanian H, Hoseinkhan N. Genetic diversity and effect of natural selection at apical membrane antigen-1 (AMA-1) among Iranian Plasmodium vivax isolates. Folia Parasitol. 2014;61(5):385–93.
Thakur A, Alam MT, Bora H, Kaur P, Sharma YD, et al. Plasmodium vivax: sequence polymorphism and effect of natural selection at apical membrane antigen 1 (PvAMA1) among Indian population. Gene. 2008;419(1–2):35–42.
Putaporntip C, Jongwutiwes S, Grynberg P, Cui L, Hughes AL, et al. Nucleotide sequence polymorphism at the apical membrane antigen-1 locus reveals population history of Plasmodium vivax in Thailand. Infect Genet Evol. 2009;9(6):1295–300.
Ord RL, Tami A, Sutherland CJ. ama1 genes of sympatric Plasmodium vivax and P. falciparum from Venezuela differ significantly in genetic diversity and recombination frequency. PLoS ONE. 2008;3(10):3366.
Genton B, Reed ZH. Asexual blood-stage malaria vaccine development: facing the challenges. Curr Opin Infect Dis. 2007;20(5):467–75.
Heidari A, Keshavarz H, Rokni MB, Jelinek T. Genetic diversity in merozoite surface protein (MSP)-1 and MSP-2 genes of Plasmodium falciparum in a major endemic region of Iran. Korean J Parasitol. 2007;45(1):59–63.
Ghanchi NK, Mårtensson A, Ursing J, Jafri S, Bereczky S, Hussain R, et al. Genetic diversity among Plasmodium falciparum field isolates in Pakistan measured with PCR genotyping of the merozoite surface protein 1 and 2. Malar J. 2010;9(1):1–6.
Paul RE, Day KP. Mating patterns of Plasmodium falciparum. Parasitol Today. 1998;14(5):197–202.
Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradović Z, et al. Intrinsic disorder and protein function. Biochemistry. 2002;41(21):6573–82.
Feng ZP, Zhang X, Han P, Arora N, Anders RF, Norton RS. Abundance of intrinsically unstructured proteins in P. falciparum and other apicomplexan parasite proteomes. Mol Biochem Parasitol. 2006;150(2):256–67.
Acknowledgements
The authors acknowledge the supportive laboratory personnel, who contributed in samples collection and diagnosis.
Funding
The study pursued without any proper grant allotment.
Author information
Authors and Affiliations
Contributions
IU, SGA and AK conceived the basic research idea. IU, MI, SS contributed in samples collection. IU, HK, KZ, AK performed data analyses. HGL, J-MK and B-KN contributed in PCR amplification and DNA sequencing experiments. IU, S.G.A and A.K prepared the initial manuscript draft. SGA, B-KN and AK finalized and reviewed the draft. All authors have agreed to the published version of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study approved from ethical review committee of Abdul Wali Khan University Mardan, Pakistan. Besides, the study pursued with informed written consent from all the study participants. All methods were carried out in accordance with relevant guidelines and regulations.
Consent for publication
Not applicable in the current study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig S1.
Frequency of different haplotypes detected in pvama-1 DI of P. vivax samples collected from different districts of KP, Pakistan
Additional file 2: Table S1.
The haplotypes identified in pvama-1 DI region of KP, Pakistan P. vivax isolates.
Additional file 3: Fig S2.
Linkage disequilibrium (LD) pattern of pvama-1 DI sequences of P. vivax isolates from KP, Pakistan. The LD index (R2) (Y-axis) plotted against nucleotide distance (X-axis) using a two tailed Fisher’s exact test.
Additional file 4: Fig S3.
Pearson correlation plot of KP and global pvama-1 samples based on pairwise Fst values. The plot shows clustering and correlation between the groups in hierarchical order. The dark brackets and large sizes depict the minimum genetic distinction and high correlation.
Additional file 5: Fig S4.
The principle component analysis (PCA) of pvama-1 DI sequences. Different colors depict different populations groups. This include KP Pakistan (as a single group) and worldwide samples.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ullah, I., Afridi, S.G., Israr, M. et al. Population genetic analyses inferred a limited genetic diversity across the pvama-1 DI domain among Plasmodium vivax isolates from Khyber Pakhtunkhwa regions of Pakistan. BMC Infect Dis 22, 807 (2022). https://doi.org/10.1186/s12879-022-07798-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-022-07798-1