
Abstract
Background
The Plasmodium falciparum apical membrane antigen-1 (PfAMA1) is considered as an ideal vaccine candidate for malaria control due to its high level of immunogenicity and essential role in parasite survival. Among the three domains of PfAMA1 protein, hyper-variable region (HVR) of domain I is the most immunogenic. The present study was conducted to evaluate the extent of genetic diversity across HVR domain I of the pfama1 gene in P. falciparum isolates from Hazara division of Pakistan.
Methods
The HVR domain I of the pfama1 was amplified and sequenced from 20 P. falciparum positive cases from Hazara division of Pakistan. The sequences were analysed in context of global population data of P. falciparum from nine malaria endemic countries. The DNA sequence reads quality assessment, reads assembling, sequences alignment/phylogenetic and population genetic analyses were performed using Staden, Lasergene v. 7.1, MEGA7 and DnaSP v.5 software packages respectively.
Results
Total 14 mutations were found in Pakistani isolates with 12 parsimony informative sites. During comparison with global isolates, a novel non-synonymous mutation (Y240F) was found specifically in a single Pakistani sample with 5% frequency. The less number of mutations, haplotypes, recombination and low pairwise nucleotide differences revealed tightly linked uniform genetic structure with low genetic diversity at HVR domain I of pfama1 among P. falciparum isolates from Hazara region of Pakistan. This uniform genetic structure may be shaped across Pakistani P. falciparum isolates by bottleneck or natural selection events.
Conclusion
The Pakistani P. falciparum isolates were found to maintain a distinct genetic pattern at HVR pfama1 with some extent of genetic relationship with geographically close Myanmar and Indian samples. However, the exact pattern of gene flow and demographic events may infer from whole genome sequence data with large sample size of P. falciparum collected from broad area of Pakistan.
Similar content being viewed by others

Background
Malaria is a vector-borne and infectious disease which is caused by a peripheral blood protozoan parasite of the genus Plasmodium [1], including five different species: Plasmodium falciparum, Plasmodium vivax, Plasmodium knowlesi, Plasmodium malariae and Plasmodium ovale [2]. The P. falciparum species accounts for the majority of the medical cases leading to lethal malaria [3, 4]. Malaria is prevalent in subtropical and tropical countries particularly Asia and Africa. In spite of advances in knowledge, the malaria disease continues to cause significant health care burden worldwide [5]. Malaria continues to have a great impact on the adults and children health all over the world. In 2016, it caused 429,000 deaths and 212 million clinical cases [5]. Malaria is considered widespread in 104 countries and territories worldwide [5]. The control and elimination for malaria is challenging due to spread of Plasmodium resistance to anti-malarial drugs along with insecticide-resistant Anopheles mosquitoes. Effective vaccine development is urgently required for better combat of malaria infection. The circumsporozoite protein (CSP), merozoite surface protein-1 (MSP-1), apical membrane antigen-1 (AMA1), and thrombospondin related anonymous protein (TRAP) are reported as vaccine candidate proteins for P. falciparum [6]. However, the genetic polymorphisms in these parasite proteins create hurdles in development of effective vaccines [7]. These polymorphisms change the critical epitopes expression and eventually reduce or cause complete loss of vaccine efficacy [8]. Therefore, extensive evaluation of genetic variants in these vaccine candidate antigenic proteins in P. falciparum populations from malaria endemic regions is primarily important for an effective and enduring vaccine development.
The AMA1 is integral membrane protein expressed in the merozoite and sporozoite stages of P. falciparum life cycle. This protein is considered to play a crucial role in invasion of erythrocytes and hepatocytes by Plasmodium [9]. The AMA1 immunization elicits antibodies production and effectively inhibits the erythrocyte invasion by the parasite [10], making AMA1 a leading vaccine candidate [11]. The AMA1 protein is comprised of three domains, and domain I exhibits high sequence polymorphism and is shown to be a key target of anti-AMA-1 protective antibodies [12]. The hyper-variable region (HVR) of domain I is highly immunogenic and natural immune responses have been reported against this domain [13]. Several studies have reported the higher rate of non-synonymous (dN) mutations at this domain due to strong diversifying selection [12, 14].
Pakistan is endemic for malaria and 60% of its population is living in malaria-endemic regions. An average 50,000 deaths occur each year in Pakistan due to malarial infection. Malaria is mostly caused by P. vivax and P. falciparum all around the Pakistan [15]. The malaria transmission remains throughout the year in Pakistan with maximum intensity from July to November [16]. The genetic structure of pfama1 gene has been reported from different malaria endemic regions of the world [12, 17, 18]. However, no report is available so far about the genetic architecture of pfama1 gene from Pakistan. The present study investigated the extent of genetic polymorphism at HVR domain I of the pfama1 among P. falciparum isolates collected from low malaria endemic Hazara division of Pakistan (i.e., termed as Pakistani Hazara, PKH).
Methods
Study area and samples collection
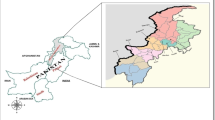
The study is carried out from Hazara division situated immediately south of the main Himalayas range in the North-Eastern part of the Khyber Pakhtunkhwa province of Pakistan (Fig. 1). Total 1961 malaria positive patients were confirmed by microscopic analysis from major hospitals of three districts (Abbottabad, Haripur and Mansehra) of Hazara division during malaria season (July–November, 2016). During data collection, 0.37 malaria cases per 1000 individuals of Hazara population were observed. According to the World Health Organization, the malaria cases > 1 per 1000 individuals of a population may consider as high transmission malaria region. The age of both male and female patients ranged from 11 to 57 years (average 28.3 years) and the infection tendency of male and female patients was found 55 and 45%, respectively in target area. Among total malaria positive cases, only 31 patients (1.6%) were diagnosed P. falciparum positive by Giemsa-stained thick smears. The blood samples (2 ml) of 20 P. falciparum positive patients were collected for DNA extraction.

Map of Pakistan, India, Myanmar and Thailand. The red dot shows the study region (i.e. Hazara division of Pakistan)
The institutional ethical guidelines were followed during blood samples collection and informed consent was obtained from all the study participants. The DNA was purified from whole blood samples using Thermo Scientific Gene JET Genomic DNA Purification Kit.
PCR amplification and DNA sequencing analysis of HVR domain I pfama1
The DNA samples were subjected to pfama1 gene HVR locus amplification through nested PCR technique using specific primers and PCR conditions reported by Garg et al. [12]. The PCR amplified products were examined through 2% agarose gel electrophoresis. The 500 bp DNA band was excised from agarose gel and purified by QIA quick Gel Extraction kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. A unidirectional Sanger-based sequencing of 500 bp purified PCR product was carried out using ABI3130 Analyzer (Applied Biosystem, USA). The cycle sequencing reactions were performed using Big Dye Terminator kit (Applied Biosystems Inc, USA) with supplier recommended conditions. The sequencing of each PCR product was performed in triplicate from single end and mutations were verified from the high-quality trace data. The sequence reads trace quality check was performed using the Staden Package [19]. The gap4 in Staden package assigns confidence values to base call which is a numerical estimate of base calling accuracy. The confidence values are stored in experiment files and copied into gap4 during data entry. All the sequence reads were evaluated on this basis and low quality reads were discarded or trimmed. Mutations were confirmed on the basis of high confidence values from triplicate (3×) sequence reads. The sequence reads assembling was performed using Lasergene package version 7.1 (DNASTAR Inc. USA). The low-quality sequences were trimmed and total 417 bp good quality sequence data was generated for each sample.
Nucleotide sequence polymorphism analysis
The nucleotide sequence polymorphism analysis of 20 Pakistani Hazara (PKH) samples was conducted. The numbers of segregating sites (S), haplotypes (H), haplotype diversity (Hd), nucleotide diversity (π), Tajima’s D, McDonald-Kreitman test, pairwise nucleotide differences within a population (K), recombination events (Rm) and Linkage disequilibrium (LD) were calculated using DnaSP v. 5 [20].
Genetic diversity analysis of HVR pfama1 among global Plasmodium falciparum isolates
The HVR pfama1 sequences of P. falciparum populations from Ghana, Tanzania, Papua New Guinea (PNG), India, Philippines, Thailand, Vanuatu and Solomon Islands were retrieved from GenBank NCBI and comparative genetic analyses were performed (Additional file 1). The samples size (n = 20) and nucleotide sequence length (i.e., 417 bp) were kept same for different parasite populations to perform unbiased statistical analysis. The allele frequencies were calculated manually on the basis of percentage prevalence of observed allele in total number of samples. The statistical analyses including genetic diversity, sequences alignment and phylogenetic analysis were conducted using DnaSP v 5 [20] and MEGA7 programs [21].
Epitope prediction
The Immune Epitope Database (IEDB) server [22] was used for PfAMA1 MHC class I and II epitopes prediction. The SMM (Stabilized matrix method) and SMM-align (netMHCII-1.1) methods were followed for human MHC class I and class II epitopes predictions respectively. The top binders with the cut-off value of IC50 < 100 nM were shortlisted to check the potential impact of the discovered nsSNPs with respect to alteration of the core sequences of lead epitopes.
Sequences submission to GenBank NCBI
The final draft of high quality DNA sequences of PKH samples were deposited to GenBank under Accession Number; MH028212-MH028193 and PopSet: 1368343752.
Results
Sequence polymorphism of PKH in HVR pfama1 sequences
The partial nucleotide sequences of 417 bp obtained for 20 PKH P. falciparum samples, spanning nucleotide positions 464–880 of reference 3D7 pfama1 sequence (Accession Number: U65407). The nucleotide sequence analysis of these sequences compared to reference revealed total 20 single nucleotide polymorphisms (SNPs), including 2 synonymous SNPs and 18 non-synonymous SNPs (nSNPs) in PKH P. falciparum isolates. These SNPs resulted in amino acid change at 15 positions in PKH samples sequences (Fig. 2). Three amino acids changes (i.e. tyrosine (Y) 175 to aspartic acid (D), lysine (K) 206 to glutamic acid (E), and isoleucine (I) 282 to lysine (K) were found fixed in all PKH samples sequences.

The polymorphic amino acid positions of P. falciparum isolates HVR pfama1 sequences from Hazara division of Pakistan. The 3D7 (GenBank Accession No.: U65407) is reference P. falciparum sequence
Population genetic analyses of PKH samples in context of global isolates
The nucleotide diversity of HVR loci at pfama1 of PKH P. falciparum isolates were analysed in context of worldwide samples deposited in GenBank (see, “Methods” section). Overall a distinct genetic structure of PKH samples were found in terms of less number of mutations, and lower values for haplotype diversity (Hd), pairwise nucleotide differences within population (K), and pairwise nucleotide diversity (π) compared to samples from other regions (Table 1). Likewise, low recombination event approaching zero and high linkage disequilibrium (LD) values were found for PKH samples compared to samples from other geographical regions (Table 2, Fig. 3). This revealed tight linkage at HVR loci for PKH samples. The LD index (R2) was found to decreases with increasing distance for target region as like most of other global samples (Fig. 3). The highest recombination events were found for African samples from Ghana and Tanzania across HVR loci as reported in previous study based on complete pfama1 gene sequences analysis [18]. The haplotype (H), Hd, K and π values calculated for PKH samples were observed close to samples from Myanmar (Table 1). In pairwise Hd and K analysis, least population-wise differences were found among Myanmar, India and PKH isolates (Table 3). This reveals close genetic relationship among geographically nearby Myanmar, India, Thailand and PKH P. falciparum isolates.

Linkage disequilibrium (LD) plot in global pfama1 including Pakistani isolates. The LD index (R2) at Y-axis was plotted against nucleotide distance across HVR pfama1 at X-axis using a two-tailed Fisher’s exact test. PKH is Pakistani Hazara, PNG is Papua New Guinea
Non-synonymous SNPs pattern of PKH in context of worldwide samples
Among non-synonymous (nsSNPs) the glycine (G) 172 to Glutamic acid (E), E187K(lysine)/N(Asparagine) and the E197Q(Glutamine)/G/D/H(Histidine) are reported as the high level of polymorphic amino acid changes in global pfama1 sequences [18]. These nsSNPs were found in PKH sequences as well. The E187K/N nsSNP was found as K187N substitution in PKH samples with major (i.e., 70%) K amino acid. Likewise at 197 position, H to D amino acid substitution was observed in PKH samples with predominant “H” amino acid at 70% frequency (Fig. 2). The Q285E was observed at low frequency (i.e. 10%) as like samples from other populations reported previously [18]. The serine (S) 283 to leucine (L) nsSNP was found with 10% frequency in PKH samples as like samples from Tanzania, Ghana, Solomon and PNG reported by Kang et al. [18]. In Myanmar and Thailand, the frequency of this nsSNP was found comparatively high, i.e. close to 40% [18]. The I (Isoleucine) 282 to K nsSNP was found fixed in PKH samples with “K” amino acid only. The high frequency of this SNP has also been reported in samples from Philippines, Ghana, and Tanzania. However the I282K frequency is reported comparatively low (i.e. < 50%) in Vanuatu, Solomon, Thailand and Myanmar [18]. The E267Q substitution was found in PKH samples with a frequency of 30%, which is more or less similar to other population samples. Only in Myanmar the frequency of E267Q substitution is reported considerably low, i.e., close to 10% [18]. The I225N, D242Y (tyrosine) and K243N substitutions were found in PKH samples with more or less same frequency as reported for other populations [18]. In some populations D242A substitution is reported, however in PKH samples no “A” substitution was found as a consequence of nsSNP at 242 residue. The K206E substitution was found fixed in PKH samples and high substitution frequency of this SNP is reported for samples from Tanzania, Ghana, Solomon, PNG, Philippines and Thailand. However, its frequency is reported comparatively low in Myanmar (i.e. < 50%) [18]. The H200D/L/R nsSNP is reported as tetra-morphic amino acid change in different continental populations, but in case of PKH samples, only D to R substitution was found at this position with predominant “D” amino acid (i.e. 70%). Likewise, the E197Q/G/D/R/H/V is reported as penta-morphic amino acid change in Myanmar and other geographical samples, however in PKH samples, it was found as H to D substitution only with predominant “H” amino acid with 70% frequency. The E187K/N nsSNP was found as K to N substitution in PKH samples with 70% frequency of “K”. The Y175D substitution was found as fixed nsSNP in PKH. This nsSNP is reported as fixed change in Myanmar pfama1 sequences as well. High frequency of Y175D substitution is reported in Africa, Oceania and East Asian samples [18]. The G172E amino acid substitution was found in PKH samples with 70% frequency of amino acid “E”. High frequency of this nsSNP is also reported in samples from Tanzania, Ghana, Thailand and Myanmar. However, in Vanuatu this amino acid substitution frequency is reported quite low, i.e., approximately 10% [18]. The N162K substitution was also found high in PKH samples with “K” amino acid frequency of 70%. This amino acid substitution frequency is reported low, i.e. close to 30% in PNG, Thailand, Myanmar and African samples from Ghana and Tanzania [18]. All the above mentioned nsSNPs identified in PKH isolates are reported in P. falciparum isolates from India. However, the detail substitution frequency of these nsSNPs is not mentioned for Indian isolates [12]. A novel Y240F substitution was found specifically in a single PKH sample (i.e., 5% frequency) during comparative sequence analysis with 459 sequences from GenBank for globally available samples. Besides, this nsSNP has not been reported in any previously published literature reports about pfama1gene analysis.
Phylogenetic analysis
The neighbour joining (NJ) method based phylogenetic analysis of PKH samples were carried out along with global pfama1 HVR sequences. Mostly the geographically distinct isolates were found to develop separate clade, however, somewhere different geographical isolates shared the same tree clade (Fig. 4). The PKH samples were found to develop two separate clusters in phylogenetic tree. However, in one major cluster the PKH samples were found to share clade with Indian, Thailand and Ghana samples (Fig. 4).

The neighbour joining phylogenetic tree constructed on the basis of HVR pfama1 sequences from PKH and global samples. The green color indicates PKH samples. Samples codes started with GHA is Ghana, TANZ is Tanzania, PHIL is Philippines, PNG is Papua New Guinea, SOL is Solomon, MYAN is Myanmar, IND is India, VAN is Vanuatu, and THAI is Thailand
Discussion
It becomes essential to investigate the antigenic repertoires of the P. falciparum populations from different malaria endemic regions. The genetic structure of Pakistani P. falciparum isolates was analysed from malaria endemic Hazara division in context of global populations based on HVR domain I of pfama1 gene. The natural selection and recombination events are previously reported as main driving forces that bring genetic diversity across pfama1 in global populations [12, 17, 18]. This adaptive evolutionary pattern of pfama1 is reported in association with host immune pressure, which eventually contributes in host immune escape [18]. The analyses of PKH samples pfama1 sequences along with worldwide P. falciparum isolates with equal samples size, result in identification of lowest haplotype number, haplotype diversity, recombination events and mutations for PKH samples as compared to global isolates (Tables 1, 2, 3). This revealed somehow the uniform genetic structure with low genetic diversity across HVR loci of pfama1 in PKH samples as compared to P. falciparum global isolates. The lower genetic diversity of PKH samples might be due to low endemicity of P. falciparum genotypes in Hazara division or might arouse due to bottleneck or natural selection. Though the Tajima’s D test result was not found statistically significant for PKH samples (Table 1), however the positive values somehow indicate the tendency of natural selection and deviation from neutral evolution as like previous studies [23,24,25]. The exact demographic event responsible for low genetic diversity of PKH isolates might be confirmed from whole genome sequence data with large samples size to be collected from broader regions of Pakistan.
The overall pattern of amino acid substitution in PfAMA1 for PKH samples were found similar to global sequences. No tri or tetra morphic amino acid changes were observed in PKH samples. However, the amino acid substitution frequencies observed for PKH samples were found different at several loci compare to samples from other malarial endemic regions. A novel Y240F amino acid substitution was observed at low frequency only in a single PKH sample. As limited number of samples is analysed in this study from a confined region of Pakistan, therefore, these amino acid changes and their frequency may not be statistically significant to represent the actual amino acid substitution spectrum of pfama1 gene across overall Pakistani region.
Most of the observed amino acid substitutions identified in our study were predicted to affect the PfAMA1 lead epitopes binding with human MHC class I and class II. Among these the H200D/R, K206E, K243N, I282K, S283L and Q285E were found to alter the PfAMA1 lead epitopes (IC50 cut off < 100 nM) cores residues and predicted to influence the epitopes binding with human host HLA-DRB1*01:01 (MHC-II). Likewise, the nsSNPs N162K, E197H/D, G172E, Y175D, I225N, I282K, S283L and Q285E were found to alter the lead epitope core sequences and suspected to influence its binding with human HLA-A*01:01 (MHC-I) allele. A novel nsSNP (i.e. Y240F) identified for a PKH sample only was not found to influence the top epitopes (i.e. IC50 cutoff < 100 nM) core sequences.
The genetic structure of HVR pfama1 gene of PKH isolates was found unique in comparison to global P. falciparum isolates sequences deposited at GenBank. During the pairwise Hd and nucleotide differences (K) analyses, the PKH isolates were found close to Myanmar samples. This might be due to close geography of PKH and Myanmar populations of the parasite. The phylogenetic tree analysis was found incongruent to this finding and the PKH and Myanmar isolates were found to develop far away independent tree clades (Fig. 4). The P. falciparum samples from India were actually collected as separate populations from different geographical regions of India as reported [12]. However, the separate population detail of these isolates is not given for DNA sequences data deposited at GenBank. Twenty of these Indian P. falciparum HVR sequences were selected to infer their genetic relationship with PKH samples. The high values of H, Hd and K for Indian samples in our analysis reflects their sub-population genetic heterogeneity. As a result, the Indian samples were found scattered throughout the phylogenetic tree and failed to develop a distinct tree clade (Fig. 4). Though Indian samples showed least difference with PKH isolates as compared to any other population in pairwise Hd and K analysis (Table 3), which reflects their close genetic affinity, however the sub-population genetic structure of Indian samples may bias such pairwise analyses. A close genetic relationship of Indian samples with PKH was also supported somehow by phylogenetic analysis as well and Indian samples were found to share a tree clade with PKH samples sequences.
Conclusion
Distinct genetic structure of pfama1 gene from P. falciparum isolates is reported from different malaria endemic regions. Therefore, a country-wide analysis is required to understand the pattern of genetic diversity across this important vaccine candidate gene. The PKH P. falciparum isolates were found to hold distinct genetic structure compared to global isolates. Overall the PKH samples exhibit uniform genetic structure with low inter-population diversity in comparison to global isolates. The population genetic analysis indicates the genetic relationship of PKH samples with geographical nearby P. falciparum isolates from Myanmar and India. Additional studies with large samples size may further validate the exact pattern of gene flow of P. falciparum isolates from Pakistani corridors into Central and South Asian regions. Likewise, the whole genome sequence data analysis will require to identify the exact demographic event responsible for low genetic diversity across HVR pfama1 in P. falciparum isolates from Pakistan.
References
Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–7.
Mohapatra PK, Prakash A, Bhattacharyya DR, Goswami BK, Ahmed A, Sarmah B, et al. Detection & molecular confirmation of a focus of Plasmodium malariae in Arunachal Pradesh, India. Indian J Med Res. 2008;128:52–6.
Urban BC, Hien TT, Day NP, Phu NH, Roberts R, Pongponratn E, et al. Fatal Plasmodium falciparum malaria causes specific patterns of splenic architectural disorganization. Infect Immun. 2005;73:1986–94.
Fu H, Hu T, Wang J, Feng D, Fang H, Wang M, et al. A bibliometric analysis of malaria research in China during 2004–2014. Malar J. 2015;14:195.
WHO. World malaria report 2016. Geneva: World Health Organization; 2016.
Richards JS, Beeson JG. The future for blood-stage vaccines against malaria. Immunol Cell Biol. 2009;87:377–90.
Takala SL, Plowe CV. Genetic diversity and malaria vaccine design, testing and efficacy: preventing and overcoming ‘vaccine resistant malaria. Parasite Immunol. 2009;31:560–73.
Srisutham S, Saralamba N, Sriprawat K, Mayxay M, Smithuis F, Nosten F, et al. Genetic diversity of three surface protein genes in Plasmodium malariae from three Asian countries. Malar J. 2018;17:24.
Yap A, Azevedo MF, Gilson PR, Weiss GE, O’Neill MT, Wilson DW, et al. Conditional expression of apical membrane antigen 1 in Plasmodium falciparum shows it is required for erythrocyte invasion by merozoites. Cell Microbiol. 2014;16:642–56.
Gentil F, Bargieri DY, Leite JA, Françoso KS, Patricio MB, Espíndola NM, et al. A recombinant vaccine based on domain II of Plasmodium vivax apical membrane antigen 1 induces high antibody titres in mice. Vaccine. 2010;28:6183–90.
Thera MA, Coulibaly D, Kone AK, Guindo AB, Traore K, Sall AH, et al. Phase 1 randomized controlled trial to evaluate the safety and immunogenicity of recombinant Pichia pastoris-expressed Plasmodium falciparum apical membrane antigen 1 (PfAMA1-FVO [25-545]) in healthy Malian adults in Bandiagara. Malar J. 2016;15:442.
Garg S, Alam MT, Das MK, Dev V, Kumar A, Dash AP, et al. Sequence diversity and natural selection at domain I of the apical membrane antigen 1 among Indian Plasmodium falciparum populations. Malar J. 2007;6:154.
Cortés A, Mellombo M, Masciantonio R, Murphy VJ, Reeder JC, Anders RF. Allele specificity of naturally acquired antibody responses against Plasmodium falciparum apical membrane antigen 1. Infect Immun. 2005;73:422–30.
Polley SD, Conway DJ. Strong diversifying selection on domains of the Plasmodium falciparum apical membrane antigen 1 gene. Genetics. 2001;158:1505–12.
Khattak AA, Venkatesan M, Nadeem MF, Satti HS, Yaqoob A, Strauss K, et al. Prevalence and distribution of human Plasmodium infection in Pakistan. Malar J. 2013;12:297.
Khan MA, Mekan SF, Abbas Z, Smego RA Jr. Concurrent malaria and enteric fever in Pakistan. Singapore Med J. 2005;46:635.
Mugyenyi CK, Elliott SR, McCallum FJ, Anders RF, Marsh K, Beeson JG. Antibodies to polymorphic invasion-inhibitory and non-inhibitory epitopes of Plasmodium falciparum apical membrane antigen 1 in human malaria. PLoS ONE. 2013;8:e68304.
Kang JM, Lee J, Moe M, Jun H, Lê HG, Kim TI, et al. Population genetic structure and natural selection of Plasmodium falciparum apical membrane antigen-1 in Myanmar isolates. Malar J. 2018;17:71.
Staden R, Beal KF, Bonfield JK. The Staden package, 1998. Methods Mol Biol. 2000;132:115–30.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;23:1451–2.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Zhang Q, Wang P, Kim Y, Haste-Andersen P, Beaver J, Bourne PE, et al. Immune epitope database analysis resource (IEDB-AR). Nucl Acids Res. 2008;36:513–8.
Cortés A, Mellombo M, Mueller I, Benet A, Reeder JC, Anders RF. Geographical structure of diversity and differences between symptomatic and asymptomatic infections for Plasmodium falciparum vaccine candidate AMA1. Infect Immun. 2003;71:1416–26.
Remarque EJ, Faber BW, Kocken CH, Thomas AW. Apical membrane antigen 1: a malaria vaccine candidate in review. Trends Parasitol. 2008;24:74–84.
Terheggen U, Drew DR, Hodder AN, Cross NJ, Mugyenyi CK, Barry AE, et al. Limited antigenic diversity of Plasmodium falciparum apical membrane antigen 1 supports the development of effective multi-allele vaccines. BMC Med. 2014;12:183.
Authors’ contributions
SGA and AK conceived and supervised the study. MI, HA and MI participated in samples collection. MA, MN and AK performed data analysis. SGA and AK wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We are thankful to Dr. Yuan Kai (Population Genomics Group, CAS-MPG Partner Institute of Computational Biology, CAS, Shanghai 200031, China) for his valuable suggestions and guidelines during the data analysis.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The raw and analysed datasets of the current study may available from the corresponding author upon request.
Consent for publication
All authors have given their consent for publication of this manuscript.
Ethics approval and consent to participate
This study was approved by the Ethical Review Committee of the Department of Biochemistry, Abdul Wali Khan University Mardan (ERC/Biochem/AWKUM/54/2016) and Center for Human Genetics, Hazara University Mansehra (CHG-HU16-016). Informed written consent form and permission were obtained from all the study participants.
Funding
This study was supported by Higher Education Commission of Pakistan under NRPU research grant award, NRPU-3474/2016. The funders had no role in study design, data collection/analyses and manuscript preparation.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1:
The NCBI accession numbers of pfama1 sequences of P. falciparum global populations used during comparative sequence analysis with PKH samples. PNG is Papua New Guinea.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Afridi, S.G., Irfan, M., Ahmad, H. et al. Population genetic structure of domain I of apical membrane antigen-1 in Plasmodium falciparum isolates from Hazara division of Pakistan. Malar J 17, 389 (2018). https://doi.org/10.1186/s12936-018-2539-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-018-2539-3