
Abstract
Colon adenocarcinoma (COAD) is one of the most frequent types of cancer worldwide. Disulfidptosis has been identified as a new mode of cell death recently. The goal of this study was to explore the possibility of a connection between disulfidptosis and COAD. RNA sequencing data from COAD patients were retrieved from the The Cancer Genome Atlas (TCGA) database for this investigation. R software and various methods were used to identify disulfidptosis-related lncRNAs (DRLs) in COAD, and a prognostic model was created based on 6 DRLs (AP003555.1, AL683813.1, SNHG7, ZEB1-AS1, AC074212.1, RPL37A-DT). The prognostic model demonstrated a good accuracy in predicting the prognosis of COAD patients, according to receiver operating characteristic (ROC) curve and Concordance index (C-index) analyses. Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed significant differences in biological functions and signaling pathways involved in differential genes in risk subgroups, including protein − DNA complex subunit organization, Hippo signaling pathway, Wnt signaling pathway. TIDE analysis was done on risk groupings in this study, and it found that patients in the high-risk group had more immune escape potential and were less probable to react to immunotherapy. Real-time quantitative pcr (qRT-PCR) was used to identify the relatively high expression of 6 DRLs in colon cancer cell lines. In summary, 6 DRLs were identified as possible novel molecular therapy targets for COAD in this investigation. This prognostic model has the potential to be a novel tool for forecasting COAD prognosis in clinical practice, as well as providing new insights on the potential function and mechanism of disulfidptosis in the COAD process.
Similar content being viewed by others

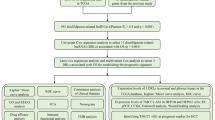

Introduction
COAD is one of the most common malignancies globally. Colon cancer has an annual average of 1,148,500 new cases and 576,800 new deaths, according to Global Cancer Statistics [1]. Because of the concealed signs of colon cancer, most patients are identified at an advanced stage. As a result, even after therapy, the prognosis for people with COAD is frequently poor [2]. In clinical practice, the prognosis of COAD patients is largely predicted by TNM staging, but the accuracy of prediction is not optimal [3]. It is critical to look for novel biomarkers to predict the prognosis of COAD patients.
By maintaining the secondary, tertiary, and quaternary structures of proteins, disulfides can improve their physical and chemical stability [4]. Liu et al. discovered a novel type of cell death known as disulfidptosis [5], which varies from other known types of death, such as necroptosis [6], pyroptosis [7], PANoptosis [8], NETosis [9]. When cells are starved, increased SLC7A11 expression causes disulfides to accumulate, resulting in disulfidptosis. The identification of a novel disulfidptosis mechanism provides new insights into the investigation of cell death.
Lncrnas have been demonstrated to have a crucial function in various kinds of malignancies. Yang et al. discovered that highly expressed LINC02159 in non-small cell lung cancer (NSCLC) tissue improved the stability of YAP1 mRNA via interacting with Aly/REF export factor (ALYREF) through m5C modification. Upregulated YAP1 can activate the Hippo and β-catenin signaling pathways, promoting NSCLC processes [10]. In artificially induced M2 human macrophages, Annika Karger et al. discovered that lncRNA ADPGK-AS1 was markedly elevated. Further research has revealed that ADPGK-AS1 is primarily found in mitochondria and binds to the mitochondrial ribosomal protein MRPL35, which promotes the tricarboxylic acid cycle and macrophage mitochondrial fission, resulting in macrophage M2 conversion and tumor growth [11]. Lncrna may be a significant prognostic indicator for COAD patients.
The relationship between lncrnas and disulfidptosis remains unclear, despite the fact that it has been demonstrated that both have a potentially significant function in COAD processes. In this work, 6 DRLs were found using integrated bioinformatics analysis and machine learning to develop a prognostic model for COAD patients. This model predicts the prognosis of COAD patients with good accuracy and is predicted to be an excellent prediction tool for forecasting the immunotherapy response and prognosis of COAD patients.
Materials and methods
Data acquisition
Four hundred seventy-six COAD tissue pairs and 41 normal tissue pairs, along with clinical data and RNA sequencing information, were acquired from the TCGA database (https://portal.gdc.cancer.gov/). Accessed 22 July 2023. Each tissue’s mRNA and lncRNA expression levels were extracted using Perl.
Identification of disulfidptosis-related lncRNAs
The expression levels of disulfidptosis-related genes (GYS1, NDUFS1, OXSM, LRPPRC, NDUFA11, NUBPL, NCKAP1, RPN1, SLC3A2, SLC7A11) in each tissue were determined by employing R software. Least absolute shrinkage and selection operator (Lasso) regression analysis and the COX model were utilized to find 6 DRLs (AP003555.1, AL683813.1, SNHG7, ZEB1-AS1, AC074212.1, RPL37A-DT) in COAD tissues based on cor ≥ 0.4 and p value ≤ 0.001.
Construction of prognostic model
The riskscore of each COAD patient was determined using the formula risk = AP003555.1x0.3279 + AL683813.1x0.5689 + SNHG7x0.5197 + ZEB1-AS1x0.4394 + AC074212.1x0.5492 + RPL37A-DTx0.8707 (the value of lncRNAs is the level of expression of lncRNAs in each patient’s tissue). Patients were divided into high-risk and low-risk categories based on the median riskscore of all COAD patients.
GO enrichment analysis and KEGG enrichment analysis
Differentially expressed genes (DEGs) in risk subgroups were identified, based on logFC ≥ 1 and fdr < 0.05. Under the condition of p value < 0.05, GO enrichment analysis and KEGG enrichment analysis of DEGs were run using the R software package, including ComplexHeatmap, ggplot2, circlize, RcolorBrewer, ggpubr, clusterProfiler, org.Hs.eg.db, enrichplot, dplyr [12].
Cell culture
From the American Type Culture Collection (ATCC), human colon cancer cell lines (SW480, SW620, LOVO) and human normal intestinal epithelial cell (FHC) were purchased. All cells were cultivated using DMEM (Gibco, C11995500BT, USA), 1640 (Gibco, C11875500BT, USA) medium in a 5% CO2, 37 °C cell incubator (Thermo, USA). All mediums included 10% foetal bovine serum (FBS) (CLARK, FB25015, USA).
RNA extraction, RNA reverse transcription and qRT-PCR
TRIZOL (Life Technologies, Thermo Fisher, USA) was used to extract total RNA from cultivated cells. cDNA was produced by reverse transcription of total RNA using the kit (Vazyme, R323-01, China). Gene expression levels were measured by LightCycler (Roche, USA) and SYBR Green Master Mix (Vazyme, Q321-02, China). Based on 2−ΔΔCT, the relative expression was computed and standardized to GAPDH expression. Primer sequence: SNHG7: LEFT PRIMER: GCAAAGAGAAAGTGGCGATT, RIGHT PRIMER: AAGTGCCCGAGCTTCAGATA; ZEB1-AS1: LEFT PRIMER: AGAAGCATCGGCTGACAGAT, RIGHT PRIMER: GGTCCCAAAGACGTTTCCTTA; RPL37A-DT: LEFT PRIMER: GCCTCCTGAGAAATGTTTGC, RIGHT PRIMER: ATGGACCCAGAGATCAATGC; AC074212.1: LEFT PRIMER: CACCTTCGGATTCCAGGAGTT, RIGHT PRIMER: CCTCCACTTGGTACTAGCTGTAAGC; AL683813.1: LEFT PRIMER: GTGCTGTTCCCTAGCACGAT, RIGHT PRIMER: CATCGAGCAGACAAGTGAGG; AP003555.1: LEFT PRIMER: CAAGGGACCACACAGGAACT, RIGHT PRIMER: GGGACATCTGGAAGCCAGT; GAPDH: LEFT PRIMER: GAAGGTGAAGGTCGGAGTC, RIGHT PRIMER: GAAGATGGTGATGGGATTTC.
Statistical analysis
All data analysis was done with R software and its tools. P < 0.05 was considered statistically significant.
Results
The identification of disulfidptosis-related lncRNAs
The expression levels of mRNAs and lncRNAs in each COAD patient were determined by downloading and re-annotating the RNA sequencing data of COAD patients (41 normal tissues and 476 COAD tissues) in the TCGA database. Further data analysis showed the expression levels of disulfidptosis-related genes (GYS1, NDUFS1, OXSM, LRPPRC, NDUFA11, NUBPL, NCKAP1, RPN1, SLC3A2, SLC7A11) in the TCGA-COAD dataset. According to the criteria of cor ≥ 0.4 and p value ≤ 0.001, disulfidptosis-related lncRNAs were screened (Fig. 1A). Lasso regression analysis and the COX model (Fig. 1B, C) were used to identify 6 DRLs for the construction of prognostic model (Fig. 1D; Table 1).

The identification of disulfidptosis-related lncRNAs. A Lncrnas associated with the disulfidptosis genes were identified in TCGA-COAD. Lasso regression analysis and the COX model (B-C) were used to identify 6 DRLs D
Construction prognostic model based on 6 disulfidptosis-related lncRNAs
Each patient’s risk score is determined using the formula of riskscore, and based on the median risk score of all patients, patients can be categorized into high risk or low risk groups. The TCGA-COAD dataset was used to acquire clinical information of patients. Patients were randomly assigned to the test group or the trian group, and there was no statistically significant clinical difference between the two groups (Table 2). The Kaplan-Meier (KM) survival analysis revealed substantial variations in overall survival (OS) between high-risk and low-risk individuals, whether in the all, train, or test groups (p<0.05) (Fig. 2A). Furthermore, there was a statistical difference between the high-risk and low-risk groups in terms of Progression Free Survival (PFS) (p<0.05) (Fig. 2B). Further investigation revealed that patients in the high-risk group had increased expression of 6 DRLs, and patient survival times were lower (Fig. 2C).

Construction and validation of prognostic model. A The KM survival analysis revealed substantial variations in OS between high-risk and low-risk individuals, whether in the all, train, or test groups. B Risk groupings’ PFS showed variation. C Patients in the high-risk group showed higher expression of 6 DRLs, and their survival times were shorter
Evaluating the prognostic model’s accuracy
Univariate and multivariate analyses revealed that age, stage, and riskScore of TCGA-COAD patients might be employed as independent prognostic indicators (Fig. 3A). ROC curve (Fig. 3B, C) and C-index analyses (Fig. 3D) revealed that the prognostic model had greater sensitivity in predicting the prognosis of COAD patients than other clinical parameters (Age, Gender, Stage). Furthermore, based on the findings of univariate analysis and multivariate analysis, we created a Nomogram by assigning age, stage, and riskScore, and the results revealed that this Nomogram could reliably predict the 1-year, 3-year, and 5-year survival rates of COAD patients (Fig. 3E). The examination of the KM survival curves revealed that the prognostic model was suitable for prognostic prediction in various clinical groups (Fig. 3F, G).

Evaluating the accuracy of the prognostic model. A Age, stage, and riskScore of TCGA-COAD patients were found to be independent prognostic markers in univariate and multivariate analyses. The prognostic model demonstrated higher sensitivity in predicting the prognosis of COAD patients, according to ROC curve (B, C) and C-index analyses (D). The 1-year, 3-year, and 5-year survival rates of COAD patients could be accurately predicted by nomogram (E). The KM survival curves demonstrated that the prognostic model was appropriate for prognostic prediction in diverse clinical groups (F, G). Pr: probability
GO enrichment analysis, KEGG enrichment analysis, immune function difference analysis and immune Escape prediction
The DEGs among risk subgroups were identified by employing differential expression analysis. According to GO enrichment analysis, the DEGs of risk categories were primarily involved in protein − DNA complex subunit organization, chromatin remodeling, structural constituent of chromatin (Fig. 4A). KEGG enrichment analysis revealed that differentially expressed genes in risk categories were mostly engaged in neutrophil extracellular trap formation, transcriptional misregulation in cancer, mammalian target of rapamycin (mTOR) signaling pathway, proteoglycans in cancer (Fig. 4B). Immune function differential study revealed variations in iDCs, Th2 cells, APC co-stimulation, CCR, and pDCs between the high-risk and low-risk groups (Fig. 4C). Furthermore, according to the TIDE prediction, the high-risk group was less responsive to immunotherapy and had a larger chance for immunological escape (Fig. 4D).

GO enrichment analysis, KEGG enrichment analysis, immune function difference analysis and TIDE analysis. GO (A) and KEGG (B) enrichment analyses revealed that DEGs in risk subgroups influenced several biological processes and cell signaling pathways of COAD. C Differential immune function analysis indicated differences between risk subgroups. D TIDE analysis found that the high-risk group was less sensitive to immunotherapy and had a higher incidence of immunological escape
Evaluating the expression levels of 6 disulfidptosis-related lncRNAs by qRT-PCR
We utilized R software to identify 6 disulfidptosis-related lncRNAs that were particularly highly expressed in COAD patients for the construction of a prognostic model to predict the prognosis of COAD patients. We utilized qRT-PCR to confirm the findings and found that AP003555.1, AL683813.1, SNHG7, ZEB1-AS1, AC074212.1 and RPL37A-DT were considerably overexpressed in human colon cancer cell lines (SW480, SW620, LOVO) compared to normal intestinal epithelial cell (FHC) (Fig. 5).

Identification of the expression level of DRLs in colon cancer cell lines
Discussion
The primary therapies for colon cancer include surgery, chemotherapy, and radiation therapy [13, 14]. Although the survival duration of certain colon cancer patients has increased due to the application of novel medicines such as molecular targeted therapy [15], immunotherapy [16, 17], and intestinal microbe management [18], many patients still have a poor prognosis. As a result, developing a novel prognostic model is beneficial in predicting the prognosis of COAD patients and subsequently developing individualized treatment for COAD patients. As a result, developing a novel prognostic model to reliably predict the prognosis of COAD patients is beneficial in developing individualized treatment for COAD patients.
Lasso regression analysis and the COX model were used in this work to identify 6 DRLs for the establishment of prognostic models, including AP003555.1, AL683813.1, SNHG7, ZEB1-AS1, AC074212.1, RPL37A-DT. Li et al. discovered that SNHG7, which is highly expressed in colorectal cancer, competes with GALNT1 for binding to miR-34a, resulting in increased GALNT7 mRNA expression. The PI3K/AKT/mTOR pathway is activated in colorectal cancer when GALNT7 is expressed at high levels, facilitating the disease spread [19]. ZEB1-AS1 is thought to play a significant role in colorectal cancer, according to several research [20, 21]. Lv et al. discovered that highly expressed ZEB1-AS1 in colorectal cancer may directly target and limit the production of miR-181a-5p, which results in increased levels of β-catenin and the transcription factor TCF4 which facilitate the development of CRC [22]. AP003555.1 has been known to be associated with ferroptosis and oxidative stress in colorectal cancer [23, 24], however there has been no investigation into the link with disulfidptosis. AC074212.1 has been identified to be involved in tumor immunity and to contribute to the prognostic prediction of endometrial carcinoma [25], but no research in colorectal cancer have been undertaken. Furthermore, the possible role and mechanism of RPL37A-DT and AL683813.1 in cancers are unknown and need to be investigated further. 6 DRLs have the potential to be novel therapeutic targets and biomarkers for colorectal cancer.
Many investigations have demonstrated that the formation and progression of malignancies is frequently associated by immune escape [26,27,28,29]. Immunotherapy is also gaining popularity in cancer treatment [30,31,32,33]. Immunotherapy can considerably enhance the overall survival of many cancer patients, but failure to respond effectively to immunotherapy in some individuals frequently leads in poor treatment outcomes [34,35,36,37,38,39]. Predicting a patient’s reaction to immunotherapy might aid in the development of more accurate therapies for patients. TIDE analysis was done on risk groupings in this study, and it found that patients in the high-risk group had more immune escape potential and were less probable to react to immunotherapy. These findings imply that this predictive model might offer some recommendations on whether COAD patients should undergo immunotherapy.
Conclusion
In general, 6 DRLs were selected for the establishment of COAD prognostic models in this study. This model shows great accuracy in predicting the prognosis of COAD patients and has the ability to provide guidance for COAD therapy in clinical practice.
Availability of data and materials
The datasets analysed during the current study are available in the TCGA databases (https://portal.gdc.cancer.gov/). Accessed 22 July 2023. In the “Materials and methods” section, the analysis methods and software applications are described. All other R code and analyses are available from the corresponding author upon request.
References
Sung H, Ferlay J, Siegel RL, et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Stoffel EM, Murphy CC. Epidemiology and mechanisms of the increasing incidence of Colon and rectal cancers in young adults. Gastroenterology. 2020;158(2):341–53.
Lea D, Håland S, Hagland HR, et al. Accuracy of TNM staging in Colorectal cancer: a review of current culprits, the modern role of morphology and stepping-stones for improvements in the molecular era. Scand J Gastroenterol. 2014;49(10):1153–63.
Zheng T, Liu Q, Xing F, et al. Disulfidptosis: a new form of programmed cell death. J Exp Clin Cancer Res. 2023;42(1):137.
Liu X, Nie L, Zhang Y, et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. 2023;25(3):404–14.
Yan J, Wan P, Choksi S, et al. Necroptosis and Tumor progression. Trends Cancer. 2022;8(1):21–7.
Yu P, Zhang X, Liu N, et al. Pyroptosis: mechanisms and Diseases. Signal Transduct Target Ther. 2021;6(1):128.
Zhu P, Ke ZR, Chen JX, et al. Advances in mechanism and regulation of PANoptosis: prospects in Disease treatment. Front Immunol. 2023;14:1120034.
Thiam HR, Wong SL, Wagner DD, et al. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020;36:191–218.
Yang Q, Wang M, Xu J, et al. LINC02159 promotes non-small cell Lung cancer progression via ALYREF/YAP1 signaling. Mol Cancer. 2023;22(1):122.
Karger A, Mansouri S, Leisegang MS, et al. ADPGK-AS1 long noncoding RNA switches macrophage metabolic and phenotypic state to promote lung cancer growth. EMBO J. 2023;42(18):e111620.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Yang Y, Wang HY, Chen YK, et al. Current status of surgical treatment of rectal cancer in China. Chin Med J (Engl). 2020;133(22):2703–11.
Dekker E, Tanis PJ, Vleugels JLA, et al. Colorectal cancer Lancet. 2019;394(10207):1467–80.
Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for Colorectal cancer. Signal Transduct Target Ther. 2020;5(1):22.
Fan A, Wang B, Wang X, et al. Immunotherapy in Colorectal cancer: current achievements and future perspective. Int J Biol Sci. 2021;17(14):3837–49.
Jiao Q, Ren Y, Ariston Gabrie AN, et al. Advances of immune checkpoints in Colorectal cancer treatment. Biomed Pharmacother. 2020;123:109745.
Montalban-Arques A, Scharl M. Intestinal microbiota and colorectal carcinoma: implications for pathogenesis, diagnosis, and therapy. EBioMedicine. 2019;48:648–55.
Li Y, Zeng C, Hu J, et al. Long non-coding RNA-SNHG7 acts as a target of miR-34a to increase GALNT7 level and regulate PI3K/Akt/mTOR pathway in Colorectal cancer progression. J Hematol Oncol. 2018;11(1):89.
Wu G, Xue M, Zhao Y, et al. Long noncoding RNA ZEB1-AS1 acts as a sponge of mir-141-3p to Inhibit Cell Proliferation in Colorectal Cancer. Int J Med Sci. 2020;17(11):1589–97.
Xiong WC, Han N, Wu N, et al. Interplay between long noncoding RNA ZEB1-AS1 and miR-101/ZEB1 axis regulates proliferation and migration of Colorectal cancer cells. Am J Transl Res. 2018;10(2):605–17.
Lv SY, Shan TD, Pan XT, et al. The lncRNA ZEB1-AS1 sponges miR-181a-5p to promote Colorectal cancer cell proliferation by regulating Wnt/β-catenin signaling. Cell Cycle. 2018;17(10):1245–54.
Wu Z, Lu Z, Li L, et al. Identification and Validation of Ferroptosis-Related LncRNA Signatures as a novel prognostic model for Colon Cancer. Front Immunol. 2022;12:783362.
Chen R, Wei JM. Integrated analysis identifies oxidative stress-related lncRNAs associated with progression and prognosis in Colorectal cancer. BMC Bioinformatics. 2023;24(1):76.
Wang Y, Liu Y, Guan Y, et al. Integrated analysis of immune-related genes in endometrial carcinoma. Cancer Cell Int. 2020;20:477.
Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to Tumor Escape. Nat Immunol. 2002;3(11):991–8.
Onkar SS, Carleton NM, Lucas PC, et al. The great Immune Escape: understanding the Divergent Immune response in Breast Cancer subtypes. Cancer Discov. 2023;13(1):23–40.
Ribas A. Adaptive Immune Resistance: how Cancer protects from Immune Attack. Cancer Discov. 2015;5(9):915–9.
Grasso CS, Giannakis M, Wells DK, et al. Genetic mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018;8(6):730–49.
Baxevanis CN, Perez SA, Papamichail M. Cancer immunotherapy. Crit Rev Clin Lab Sci. 2009;46(4):167–89.
Borgers JSW, Heimovaara JH, Cardonick E, et al. Immunotherapy for cancer treatment during pregnancy. Lancet Oncol. 2021;22(12):e550–61.
Yu WD, Sun G, Li J, et al. Mechanisms and therapeutic potentials of cancer immunotherapy in combination with radiotherapy and/or chemotherapy. Cancer Lett. 2019;452:66–70.
Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17(8):807–21.
Emens LA. Breast Cancer Immunotherapy: facts and hopes. Clin Cancer Res. 2018;24(3):511–20.
Tiwari A, Trivedi R, Lin SY. Tumor microenvironment: barrier or opportunity towards effective cancer therapy. J Biomed Sci. 2022;29(1):83.
Weng J, Li S, Zhu Z, et al. Exploring immunotherapy in Colorectal cancer. J Hematol Oncol. 2022;15(1):95.
Schuster SJ, Tam CS, Borchmann P, et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell Lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021;22(10):1403–15.
Fowler NH, Dickinson M, Dreyling M, et al. Tisagenlecleucel in adult relapsed or refractory follicular Lymphoma: the phase 2 ELARA trial. Nat Med. 2022;28(2):325–32.
Takeda Y, Kataoka K, Yamagishi J, et al. A TLR3-Specific adjuvant relieves Innate Resistance to PD-L1 Blockade without Cytokine Toxicity in Tumor Vaccine Immunotherapy. Cell Rep. 2017;19(9):1874–87.
Funding
This research received no external funding.
Author information
Authors and Affiliations
Contributions
CP and YJ performed the data analysis. YJ and LQ designed this work. CP and LJ wrote the manuscript. LJ and WW verified the conclusion through the experiment . All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, P., Yu, J., Luo, Q. et al. Construction of disulfidptosis-related lncRNA signature for predicting the prognosis and immune escape in colon adenocarcinoma. BMC Gastroenterol 23, 382 (2023). https://doi.org/10.1186/s12876-023-03020-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-023-03020-x