
Abstract
Background
Accumulating studies have demonstrated that lncRNAs play vital roles in the prognosis of gastric cancer (GC); however, the prognostic value of N6-methyladenosine-related lncRNAs has not been fully reported in GC. This study aimed to construct and validate an m6A-related lncRNA pair signature (m6A-LPS) for predicting the prognosis of GC patients.
Methods
GC cohort primary data were downloaded from The Cancer Genome Atlas. We analysed the coexpression of m6A regulators and lncRNAs to identify m6A-related lncRNAs. Based on cyclical single pairing along with a 0-or-1 matrix and least absolute shrinkage and selection operator-penalized regression analyses, we constructed a novel prognostic signature of m6A-related lncRNA pairs with no dependence upon specific lncRNA expression levels. All patients were divided into high-risk and low-risk group based on the median risk score. The predictive reliability was evaluated in the testing dataset and whole dataset with receiver operating characteristic (ROC) curve analysis. Gene set enrichment analysis was used to identify potential pathways.
Results
Fourteen m6A-related lncRNA pairs consisting of 25 unique lncRNAs were used to construct the m6A-LPS. Kaplan–Meier analysis showed that the high-risk group had poor prognosis. The area under the curve for 5-year overall survival was 0.906, 0.827, and 0.882 in the training dataset, testing dataset, and whole dataset, respectively, meaning that the m6A-LPS was highly accurate in predicting GC patient prognosis. The m6A-LPS served as an independent prognostic factor for GC patients after adjusting for other clinical factors (p < 0.05). The m6A-LPS had more accuracy and a higher ROC value than other prognostic models for GC. Functional analysis revealed that high-risk group samples mainly showed enrichment of extracellular matrix receptor interactions and focal adhesion. Moreover, N-cadherin and vimentin, known biomarkers of epithelial–mesenchymal transition, were highly expressed in high-risk group samples. The immune infiltration analysis showed that resting dendritic cells, monocytes, and resting memory CD4 T cells were significantly positively related to the risk score. Thus, m6A-LPS reflected the infiltration of several types of immune cells.
Conclusions
The signature established by pairing m6A-related lncRNAs regardless of expression levels showed high and independent clinical prediction value in GC patients.
Similar content being viewed by others

Introduction
Gastric cancer (GC) is a major global disease, and it is the fifth most common cancer and the fourth most lethal malignancy. There were more than one million new cases and an estimated 769,000 deaths in 2020 [1], and more than 40% of the new cases and deaths occurred in China [2, 3]. In addition, 80% of patients with GC are diagnosed at an advanced stage [4]. Notably, the 5-year mortality rate for advanced GC is between 30 and 50% [5]. Overall, the prognosis of GC is not very optimistic, and it is necessary to identify novel biomarkers to reliably predict the survival outcomes of GC patients.
Of the over 160 RNA post-transcriptional regulatory marks in multiple RNA species, N6-methyladenosine (m6A) is the most common form modification on mRNA in higher eukaryotes, and it plays a vital role in RNA splicing, export, stability and translation [6]. Recently, accumulating studies have revealed that m6A modification is involved in multiple processes of tumorigenesis [7,8,9,10,11], and m6A modification, which is a reversible and dynamic process, is regulated by m6A regulators, including “writers” (methyltransferases), “readers” (signal transducers) and “erasers” (demethylases) [12]. Writers, including METTL3, METTL16, KIAA1429, WTAP, RBM15, RBM15B, and ZC3H13, mediate the RNA methylation modification process. Erasers include FTO and ALKBH5, and mediate the RNA demethylation process. In addition, signal transducers, including YT521-B homology (YTH) domain family members (YTHDF1, YTHDF2, and YTHDF3), YTH domain-containing proteins (YTHDC1 and YTHDC2), heterogeneous nuclear ribonucleoproteins family members (HNRNP and HNRNPA2B1), and insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs; including IGF2BP1, IGF2BP2, and IGF2BP3), affect the reading of RNA methylation information, translation, stability and degradation of downstream RNAs [4, 13, 14]. In summary, m6A RNA methylation has a significant impact on RNA production and metabolism and is involved in the pathogenesis of multiple diseases, including GC [15].
Long non-coding RNAs (lncRNAs) represent the largest group of non-coding RNAs produced from the genome [16], and they are more than 200 nucleotides in length. Accumulating evidence has revealed that various lncRNAs contribute to gene expression at both the post-transcriptional and transcriptional levels. Additionally, aberrant lncRNA expression is strongly related to multiple cancers [12, 17] and serves as a diagnostic and prognostic marker for tumours [18]. Furthermore, lncRNAs can direct the expression of genes related to the activation of immune cells, thus altering the immune microenvironment and further contributing to the malignant phenotypes of some cancers [17, 19]. m6A-related lncRNAs are potential biomarkers for predicting the overall survival (OS) of lower-grade glioma patients and might be novel therapeutic targets [12]. However, m6A-related lncRNA signatures in GC patients need further exploration.
Epithelial–mesenchymal transition (EMT) is a process that enables polarized epithelial cells to transition towards a mesenchymal phenotype with increased cellular motility, and EMT occurs in many types of cancers [20]. In GC, the loss of E-cadherin expression stimulates cell transformation into a more invasive and less differentiated state through the EMT process [21]. However, the association between m6A-related lncRNAs and EMT factors in GC is not entirely clear.
In the present study, we analysed the value of a m6A-related lncRNA pair signature (m6A-LPS) in predicting the OS of GC patients and further validated the m6A-LPS in the testing dataset and the whole dataset. Notably, m6A-LPS served as an independent prognostic marker for GC independent of other clinical variables. Additionally, we identified differences in the expression of EMT biomarkers and immune cell infiltration between the high-risk and low-risk groups.
Materials and methods
Data collection and preparation, correlation analysis and differential expression analysis
All data, including the RNA-seq reads per kilobase per million (FPKM) data and clinical information of GC samples, were downloaded from The Cancer Genome Atlas (TCGA) database. By using GTF file annotation, mRNAs and lncRNAs were distinguished. m6A-related lncRNAs were defined as those with Pearson correlation coefficient > 0.4 and p < 0.001. Additionally, differential expression analysis of m6A-related lncRNAs between normal and adjacent tissue was performed using the R package limma, including thresholds of |log fold change (FC)| > 1.5 and false discovery rate (FDR) < 0.05.
lncRNA pairs
The differentially expressed m6A-related lncRNAs were cyclically single paired, and a lncRNA pair matrix was constructed. Briefly, if the expression level of the first lncRNA was higher than that of the second lncRNA, the expression was assigned as 1; otherwise, the output was 0. In addition, the lncRNA pair was identified as a valid match when the number of pairs with an expression quantity of 0 or 1 accounted for more than 20% of the total lncRNA pairs.
Construction of a m6A-LPS and evaluation of the relative risk score
First, we utilized univariate survival analysis based on the Kaplan–Meier method with the log-rank test to identify prognostic m6A-related lncRNA pairs, and a p value < 0.05 was considered to indicate statistical significance. To avoid overfitting, least absolute shrinkage and selection operator (LASSO)-penalized regression analysis was used to construct the best model. The following formula was used to calculate the risk score of each GC patient.
where “n” means the total number of lncRNA pairs included in the signature, “Expr” is the matrix value of the lncRNA (either 1 or 0), and “Coef” is the coefficient of the lncRNA pair estimated from the LASSO regression model. All of the GC patients were randomly divided into a training dataset and a testing dataset. Then, the patients were divided into a high-risk group and a low-risk group based on the median risk score. Kaplan–Meier analysis and ROC curve analysis were used to evaluate the OS prediction ability and prognostic accuracy of m6A-LPS in the training dataset, the testing dataset, and the whole dataset. The sensitivity and specificity of m6A-LPS for GC patients was compared with those of other clinicopathological characteristics using ROC curve analysis and decision curve analysis (DCA) [22].
Validation of the model and predictive nomogram
The chi-square test was used to confirm the relationship between the m6A-LPS and clinicopathological characteristics, and univariate and multivariate Cox regression analyses were used to determine whether the m6A-LPS was an independent prognostic predictor. Kaplan–Meier analysis was used to confirm the predictive value of the risk score in different clinicopathological feature subgroups. Additionally, a nomogram was constructed by integrating the m6A-LPS and clinicopathological features to predict the 1-, 3-, and 5-year OS of GC patients.
Investigation of tumour-infiltrating immune cells
We used CIBERSORT to analyse the relationship between the risk score and immune cells. The relationships were analysed by Spearman correlation analysis, and p < 0.05 was considered to indicate statistical significance. The procedure used the R ggplot 2 package.
Gene set enrichment analysis (GSEA)
GSEA was used to quantify the underlying Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with the m6A-LPS, and p < 0.05 and FDR < 0.05 were used as the criteria to identify significant pathways.
Statistical analysis
All primary data were downloaded from TCGA, and all statistical analyses were performed using R (version 4.0.4) and PERL (version 5.32.1). Survival differences were determined using Kaplan–Meier curve and log-rank test analyses, and the survival curves were plotted with the R package survmine. Multivariate analyses were conducted using the Cox proportional hazard regression model. Clinical data were analysed using the chi-square test or Fisher’s exact test. For all results, a p value < 0.05 was considered to indicate statistical significance.
Result
Identification of differentially expressed m6A-related lncRNAs
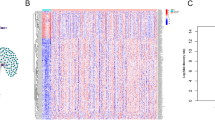
The transcriptome profiling data of GC samples, including 32 adjacent and 375 tumour tissue samples, were downloaded from TCGA. We identified 14,086 lncRNAs in the GC dataset. A total of 23 m6A regulators were acquired from published studies (Table 1), and 10 of 23 m6A regulators with hazard ratio (HR) > 1 and p < 0.05 in GC patients were further screened in Kaplan–Meier Plotter (Table 2). Their expression in GC patients is shown in Fig. 1. Heatmap analysis showed that 10 m6A regulators were significantly more highly expressed in tumour tissue than in normal tissue (p < 0.05), except for FTO and ALKBH5. Furthermore, 491 lncRNAs related to 23 m6A regulators were identified, and 444 m6A-related lncRNAs were further selected based on 10 m6A regulators for the next part of the study. A total of 85 differentially expressed m6A-related lncRNAs were identified (Fig. 2a), with filter conditions of| log FC| > 1.5 and FDR < 0.05; of these, 60 were upregulated and 25 were downregulated (Fig. 2b).

Expression of the 10 m6A regulators in GC patients. (***p < 0.001, **p < 0.01, *p < 0.05)

The 85 differently expressed lncRNAs in GC patients (a heatmap; b volcano plot)
Establishment of m6A-related lncRNA pairs and a risk assessment model
First, among the 85 differentially expressed m6A-related lncRNAs, 2069 valid m6A-related lncRNA pairs were identified by using an iteration loop and a 0-or-1 matrix. We randomly divided 347 patients into a training dataset (N = 174) and a testing dataset (N = 173) (training dataset: test dataset = 1:1). We then used univariate Cox regression analysis and LASSO-penalized regression analysis to construct the m6A-LPS in the training dataset (Fig. 3). Finally, we identified 14 m6A-related lncRNA pairs and their corresponding coefficients (Table 3). The risk scores of each patient in the training dataset, testing dataset, and the whole dataset were calculated based on the following risk formula:

Characterization of the m6A-LPS. a LASSO coefficients of the 14 lncRNA pairs in GC patients. b Selection of the best parameters for GC patients on the basis of the LASSO model
Patients in the three datasets were further divided into a high-risk group and a low-risk group based on the median risk score. The Kaplan–Meier curve analysis results showed that the low-risk group had a better prognosis than the high-risk group in the three datasets (p < 0.001) (Fig. 4a–c). Moreover, the area under the curve (AUC) for 5-year OS was 0.906, 0.827, and 0.882 in the training dataset, testing dataset, and whole dataset, respectively (Fig. 4d–f). Furthermore, the AUC of m6A-LPS was 0.882, exhibiting superior performance compared to traditional clinicopathological characteristics in predicting the prognosis of GC patients (Fig. 4g–h).

Kaplan–Meier survival curve (a–c) and ROC curve (d–f) analysis of the m6A-LPS between the high-risk group and low-risk group in the training dataset (a, d), testing dataset (b, e), and whole dataset (c, f). g Comparison of the 5-year ROC curves of the m6A-LPS and other clinicopathological features. h DCA of the risk factors
Prognostic value of m6A-LPS and its relationship with clinicopathological features
The distribution of m6A-LPS was plotted along with the corresponding survival status based on the risk curve in Fig. 5a–c. The results showed that as the risk score increased, the number of deaths and the proportion of high-risk patients increased in the three datasets. Subsequently, to verify the clinical application value of the m6A-LPS, we performed univariate Cox regression analysis and multivariate Cox regression analysis of the m6A-LPS and clinicopathological characteristics, such as age, sex, grade and stage. The results revealed that m6A-LPS was an independent factor for predicting the prognosis of GC (p < 0.001) (Fig. 5d, e). Finally, to determine the predictive value of the m6A-LPS in different clinicopathological feature subgroups, we performed a stratified survival analysis. The survival curve revealed that m6A-LPS was a stable prognostic marker (p < 0.001) for GC patients stratified by age (<= 65 or > 65), sex (male or female), grade (G1-2 or G3), and stage (I–II or III–IV), as shown in Fig. 6. Furthermore, we compared our m6A-LPS with three published representative gene prognostic markers [23,24,25] using ROC curves for 1-, 3-, and 5-year OS, as shown in Fig. 7. The results showed that the 5-year AUC value of our prognostic model (the m6A-LPS) was 0.882, showing obviously higher predictive value and accuracy than the existing prognostic models Lv.signature (5-year AUC = 0.630), Liu.signature (5-year AUC = 0.675), and Mao. signature (5-year AUC = 0.577). Finally, the hybrid nomogram incorporating clinicopathological features and the m6A-LPS was also found to be stable and accurate, suggesting that it has potential value in the clinical management of GC patients (Fig. 8).

The distributions of the m6A-LPS along with the corresponding survival status based on the risk curve and the independence of the m6A-LPS in OS. (a the training dataset, b testing dataset, c whole dataset, d Univariate cox regression analysis, e Multivariate cox regression analysis)

Kaplan–Meier survival curves for the high-risk and low-risk groups stratified by clinical factors including age (a, b), sex (c, d), grade (e, f), and stage (g, h) in the whole dataset

ROC analysis of different prognostic signatures. The 5-year overall survival AUC value of m6A-LPS model, Lv.signature model, Liu.signature model and Mao.signature model were 0.882, 0.630, 0.675 and 0.577, respectively

A nomogram including both clinicopathological factors and the m6A-LPS (**p < 0.05, ***p < 0.001)
GSEA
GSEA was used to explore the potential functions or pathways of the m6A-LPS. We defined the high-risk group as cluster 2 and the low-risk group as cluster 1. We found that patients in the high-risk group mainly showed enrichment of the terms extracellular matrix (ECM) receptor interactions and focal adhesion, while the low-risk group was characterized by enriched homologous recombination, oxidative phosphorylation and base excision repair (Fig. 9).

Gene set enrichment analysis of the m6A-LPS in the high-risk group (a, b) and low-risk group (c, d)
Association between tumour-infiltrating immune cells and the prognostic model
To explore the potential relationship between m6A-LPS and infiltrating immune cells, the Wilcoxon signed-rank test was utilized. The results revealed that monocytes (R = 0.18, p = 0.0095), M2 macrophages (R = 0.15, p = 0.034), resting dendritic cells (R = 0.15, p = 0.0029), and resting memory CD4 T cells (R = 0.16, p = 0.017) were positively correlated with the risk score, while activated memory CD4 T cells (R = − 0.14, p = 0.044) were inversely correlated with the risk score in the CIBERSORT dataset (Fig. 10).

Potential relationships between the m6A-LPS and infiltrating immune cells
EMT biomarkers
Increasing evidence has revealed that EMT is the basis of invasion and metastatic cancer cell spreading [20]. Therefore, we further determined EMT biomarkers that were differentially expressed in the high-risk and low-risk groups, and the results showed that N-cadherin and vimentin, which are markers of mesenchymal cells, were highly expressed in the high-risk group (p < 0.05); however, E-cadherin, a marker of epithelial cells, was not significantly different between the groups (p > 0.05) (Fig. 11).

EMT biomarkers differentially expressed between the high-risk and low-risk groups
Discussion
With the rapid development of high-throughput sequencing and bioinformatics analyses, we are entering a new era of biological big data. A tremendous amount of genomic information, including potential biomarkers, can be detected in clinical samples, promoting the diagnosis, prognostication and prediction of diseases [26]. Genomic signatures are novel biomarkers in which genomic data are combined in a defined manner and have been proven to be able to predict the prognosis of patients with diseases, especially those with malignant tumours [27]. GC remains one of the most prevalent and deadly cancers worldwide, especially in China. Due to the lack of diagnostic biomarkers, most patients are diagnosed at an advanced stage, and not all patients benefit equally from surgical resection, chemotherapy or chemoradiotherapy because of disease heterogeneity [28]. In recent years, an increasing number of studies have focused on establishing signatures with both coding genes and noncoding RNAs to evaluate the prognosis of patients with cancer [29]. Several studies have revealed that m6A-related lncRNAs participate in the development of various cancers, including GC. Thus, exploring the role of lncRNAs in the prognosis and diagnosis of GC will contribute to a better understanding of the molecular mechanism of GC [13]. However, most prognostic signatures published to date [13, 23,24,25, 30, 31] require proper standardization of gene expression profile data for further analysis, which is a major limitation in clinical application. In the current study, we employed a strategy considering immune-related gene pairs [28] and attempted to construct an efficient model with two-lncRNA combinations regardless of exact expression levels [32]; this strategy not only eliminates batch effects among different platforms but also lacks the need for the normalization and scaling of data, thus successfully solving the problems surrounding the use of different data platforms to determine expression [33, 34].
First, raw lncRNA data were downloaded from the GC project of TCGA. By performing Pearson correlation coefficient analysis, iteration loop, 0-or-1 matrix, univariate Cox regression and LASSO-penalized regression analyses, we constructed an m6A-LPS (containing 14 m6A-related lncRNA pairs consisting of 25 unique lncRNAs). Based on the median risk score, patients were divided into high-risk and low-risk groups, and Kaplan–Meier curve analysis revealed that the high-risk group had shorter OS. Further ROC analysis revealed that the m6A-LPS had a higher accuracy in predicting the 5-year OS of GC than other traditional clinicopathological features. Moreover, multivariate Cox regression analysis revealed that m6A-LPS was an independent risk factor for GC. Notably, we also compared the accuracy of our model with that of other reported models. The AUC values of the Lv et al. seven-mRNA signature in predicting the 1-, 3-, and 5-year OS were 0.682, 0.603, and 0.630, respectively, and the AUC values of the Liu et al. four-gene signature in predicting the 1-, 3-, and 5-year OS were 0.535, 0.617, and 0.675, respectively. The AUC values of the Mao et al. six-gene signature in predicting the 1-, 3-, and 5-year OS were 0.557, 0.615, and 0.577, respectively, while the AUCs for our m6A-LPS model in predicting the OS at 1, 3, and 5 years were 0.795, 0.818, and 0.882, respectively. All of the above results demonstrate that our m6A-LPS provides efficient and robust prognostic prediction and might serve as an efficient biomarker for the prognosis of GC. In addition, a nomogram based on the m6A-LPS and clinicopathological factors may be applied in the clinical management of GC patients.
Furthermore, the GSEA results showed that patients in the high-risk group mainly showed enrichment of ECM receptor interactions and focal adhesion. Notably, previous studies have demonstrated that the ECM plays a vital role in cancer progression, and focal adhesion kinase (FAK) is often associated with poor clinical outcome, highlighting FAK as a potential determinant of tumour progression and metastasis [35]. The above results provide new directions for exploring the potential molecular mechanisms of GC.
Moreover, previous studies revealed that tumour-infiltrating immune cells can be used as independent prognostic markers in GC [36]. Therefore, we used CIBERSORT to explore the relationship between the risk score and tumour-infiltrating immune cells. The results showed that resting memory CD4 T cells, resting dendritic cells, monocytes, and M2 macrophages were positively related to the risk score, while activated memory CD4 T cells were inversely correlated with the risk score. Published studies have shown that increased monocytes and activated memory CD4 T cells are related to the poor prognosis of GC [37, 38], which is consistent with our research.
Finally, we also analysed the differential expression of EMT biomarkers between the high-risk and low-risk groups because the EMT process is a key molecular step in distant metastasis and is associated with poor prognosis [39]. The results showed that N-cadherin and vimentin, biomarkers of mesenchymal cells, were abundantly expressed in the high-risk group of patients. These results may provide new ideas for individualized treatment of GC patients.
Overall, we developed a prognostic model based on 14 m6A-related lncRNA pairs, and only the relative expression of the pairs had to be detected instead of examining specific expression values of every lncRNA, significantly lowering the cost of sequencing and carrying high clinical practicability. Furthermore, the prognostic model showed a robust, high value for predicting the survival of GC. However, this study has several limitations that need to be addressed. First, our prognostic model was constructed based only on TCGA data, and we failed to use other public databases or patient cohorts for further validation. Second, the relationship between m6A regulators and lncRNAs should be further explored in experiments in vitro and in vivo.
Conclusion
In the current study, we constructed an m6A-LPS prognostic model with high predictive value that can serve as an independent prognostic factor for GC. To the best of our knowledge, this is the first study to construct a prognostic model based on m6A-related lncRNA pairs that does not require assessment of the exact expression levels of each lncRNA. Obviously, it has substantial value in clinical applications. Additionally, our results provide a new direction for individualized therapy.
Availability of data and materials
The raw data of this study are derived from the TCGA database (https://portal.gdc.cancer.gov/) and Kaplan–Meier Plotter data portal (http://kmplot.com/analysis/), which are publicly available databases.
Abbreviations
- m6A:
-
N6-methyladenosine
- GC:
-
Gastric cancer
- m6A-LPS:
-
M6A-related lncRNA pairs signature
- TCGA:
-
The cancer genome atlas program
- LASSO:
-
The least absolute shrinkage and selection operator
- ROC:
-
The receiver operating characteristic curve
- AUC:
-
The area under the curve
- DCA:
-
The decision curve analysis
- LncRNA:
-
Long non coding RNA
- EMT:
-
Epithelial mesenchymal cell transition
- HR:
-
Hazard ratio
- FC:
-
Fold change
- FDR:
-
False discovery rate
- OS:
-
Overall survival
- GSEA:
-
Gene set enrichment analysis
- KEGG:
-
Kyoto enrcyclopedia of genes and genomes
- KM:
-
The Kaplan–Meier
- ECM:
-
Extracellular matrix
- FAK:
-
The focal adhesion kinase
References
Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021. https://doi.org/10.3322/caac.21660.
Jin G, Lv J, Yang M, et al. Genetic risk, incident gastric cancer, and healthy lifestyle: a meta-analysis of genome-wide association studies and prospective cohort study. Lancet Oncol. 2020. https://doi.org/10.1016/S1470-2045(20)30460-5.
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016. https://doi.org/10.3322/caac.21338.
Lordick F, Allum W, Carneiro F, et al. Unmet needs and challenges in gastric cancer: the way forwardsforward. Cancer Treat Rev. 2014. https://doi.org/10.1016/j.ctrv.2014.03.002.
Katai H, Ishikawa T, Akazawa K, et al. Five-year survival analysis of surgically resected gastric cancer cases in Japan: a retrospective analysis of more than 100,000 patients from the nationwide registry of the Japanese Gastric Cancer Association (2001–2007). Gastric Cancer. 2018. https://doi.org/10.1007/s10120-017-0716-7.
Sang L, Sun L, Wang A, et al. The N6-methyladenosine features of mRNA and aberrant expression of m6A modified genes in gastric cancer and their potential impact on the risk and prognosis. Front Genet. 2020. https://doi.org/10.3389/fgene.2020.561566.
Dai D, Wang H, Zhu L, et al. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018. https://doi.org/10.1038/s41419-017-0129-x.
Zhou Y, Yin Z, Hou B, et al. Expression profiles and prognostic significance of RNA N6-methyladenosine-related genes in patients with hepatocellular carcinoma: evidence from independent datasets. Cancer Manag Res. 2019. https://doi.org/10.2147/CMAR.S191565.
Han J, Wang JZ, Yang X, et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. 2019. https://doi.org/10.1186/s12943-019-1036-9.
Li T, Hu PS, Zuo Z, et al. METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019. https://doi.org/10.1186/s12943-019-1038-7.
Yi L, Wu G, Guo L, et al. Comprehensive analysis of the PD-L1 and immune infiltrates of m6A RNA methylation regulators in head and neck squamous cell carcinoma. Mol Ther Nucleic Acids. 2020. https://doi.org/10.1016/j.omtn.2020.06.001.
Tu Z, Wu L, Wang P, et al. N6-methylandenosine-related lncRNAs are potential biomarkers for predicting the overall survival of lower-grade glioma patients. Front Cell Dev Biol. 2020. https://doi.org/10.3389/fcell.2020.00642.
Wang H, Meng Q, Ma B. Characterization of the prognostic m6A-related lncRNA signature in gastric cancer. Front Oncol. 2021. https://doi.org/10.3389/fonc.2021.630260.
Huang H, Weng H, Sun W, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018. https://doi.org/10.1038/s41556-018-0045-z.
Chen XY, Zhang J, Zhu JS. The role of m6A RNA methylation in human cancer. Mol Cancer. 2019. https://doi.org/10.1186/s12943-019-1033-z.
Robinson EK, Covarrubias S, Carpenter S. The how and why of lncRNA function: an innate immune perspective. Biochim Biophys Acta Gene Regul Mech. 2020. https://doi.org/10.1016/j.bbagrm.2019.194419.
Atianand MK, Caffrey DR, Fitzgerald KA. Immunobiology of long noncoding RNAs. Annu Rev Immunol. 2017. https://doi.org/10.1146/annurev-immunol-041015-055459.
Poursheikhani A, Abbaszadegan MR, Nokhandani N, et al. Integration analysis of long non-coding RNA (lncRNA) role in tumorigenesis of colon adenocarcinoma. BMC Med Genom. 2020. https://doi.org/10.1186/s12920-020-00757-2.
Chen YG, Satpathy AT, Chang HY. Gene regulation in the immune system by long noncoding RNAs. Nat Immunol. 2017. https://doi.org/10.1038/ni.3771.
Lu W, Kang Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev Cell. 2019. https://doi.org/10.1016/j.devcel.2019.04.010.
Bure IV, Nemtsova MV, Zaletaev DV. Roles of E-cadherin and noncoding RNAs in the epithelial-mesenchymal transition and progression in gastric cancer. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20122870.
Vickers AJ, Elkin EB. Decision curve analysis: a novel method for evaluating prediction models. Med Decis Making. 2006. https://doi.org/10.1177/0272989X06295361.
Lv X, Zhao Y, Zhang L, et al. Development of a novel gene signature in patients without Helicobacter pylori infection gastric cancer. J Cell Biochem. 2020. https://doi.org/10.1002/jcb.29419.
Liu Y, Wu J, Huang W, et al. Development and validation of a hypoxia-immune-based microenvironment gene signature for risk stratification in gastric cancer. J Transl Med. 2020. https://doi.org/10.1186/s12967-020-02366-0.
Mao R, Wang Z, Zhang Y, et al. Development and validation of a novel prognostic signature in gastric adenocarcinoma. Aging (Albany NY). 2020. https://doi.org/10.18632/aging.104161.
Subramanian J, Simon R. What should physicians look for in evaluating prognostic gene-expression signatures? Nat Rev Clin Oncol. 2010. https://doi.org/10.1038/nrclinonc.2010.60.
Peng PL, Zhou XY, Yi GD, et al. Identification of a novel gene pairs signature in the prognosis of gastric cancer. Cancer Med. 2018. https://doi.org/10.1002/cam4.1303.
Zhao E, Zhou C, Chen S. A signature of 14 immune-related gene pairs predicts overall survival in gastric cancer. Clin Transl Oncol. 2021. https://doi.org/10.1007/s12094-020-02414-7.
Zhu X, Tian X, Yu C, et al. A long non-coding RNA signature to improve prognosis prediction of gastric cancer. Mol Cancer. 2016. https://doi.org/10.1186/s12943-016-0544-0.
Liu X, Wu J, Zhang D, et al. Identification of potential key genes associated with the pathogenesis and prognosis of gastric cancer based on integrated bioinformatics analysis. Front Genet. 2018. https://doi.org/10.3389/fgene.2018.00265.
Jiang B, Sun Q, Tong Y, et al. An immune-related gene signature predicts prognosis of gastric cancer. Medicine (Baltimore). 2019. https://doi.org/10.1097/MD.0000000000016273.
Hong W, Liang L, Gu Y, et al. Immune-related lncRNA to construct novel signature and predict the immune landscape of human hepatocellular carcinoma. Mol Ther Nucleic Acids. 2020. https://doi.org/10.1016/j.omtn.2020.10.002.
Qi L, Chen L, Li Y, et al. Critical limitations of prognostic signatures based on risk scores summarized from gene expression levels: a case study for resected stage I non-small-cell lung cancer. Brief Bioinform. 2016. https://doi.org/10.1093/bib/bbv064.
Geman D, d’Avignon C, Naiman DQ, Winslow RL. Classifying gene expression profiles from pairwise mRNA comparisons. Stat Appl Genet Mol Biol. 2004. https://doi.org/10.2202/1544-6115.1071.
Lee BY, Timpson P, Horvath LG, et al. FAK signaling in human cancer as a target for therapeutics. Pharmacol Ther. 2015. https://doi.org/10.1016/j.pharmthera.2014.10.001.
Walker C, Mojares E, Del Río Hernández A. Role of extracellular matrix in development and cancer progression. Int J Mol Sci. 2018. https://doi.org/10.3390/ijms19103028.
Zeng D, Zhou R, Yu Y, et al. Gene expression profiles for a prognostic immunoscore in gastric cancer. Br J Surg. 2018. https://doi.org/10.1002/bjs.10871.
Urakawa S, Yamasaki M, Goto K, et al. Peri-operative monocyte count is a marker of poor prognosis in gastric cancer: increased monocytes are a characteristic of myeloid-derived suppressor cells. Cancer Immunol Immunother. 2019. https://doi.org/10.1007/s00262-019-02366-0.
Yue B, Song C, Yang L, et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. 2019. https://doi.org/10.1186/s12943-019-1065-4.
Acknowledgements
The authors gratefully acknowledge the Cancer Genome Atlas (TCGA) database,and the Kaplan–Meier Plotter, which made the data available.
Funding
This study was supported by the International Science and technology cooperation fund of Changzhou, China (CZ20190021), Young medical talents of Jiangsu province (QNRC2016269), Changzhou special fund for introducing foreign talents (CQ20204037), and the Jiangsu postdoctoral fund.
Author information
Authors and Affiliations
Contributions
WXY and WJM designed the study, WJM wrote the manuscript, YP and GWB contributed to collect the resourses. WXY, LX contributed to reviewing the draft and final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of The Cancer Genome Atlas Human Subjects Protection and Data Access Policies, adopted by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI). Informed consent was obtained from all individual participants included in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, JM., Li, X., Yang, P. et al. Identification of a novel m6A-related lncRNA pair signature for predicting the prognosis of gastric cancer patients. BMC Gastroenterol 22, 76 (2022). https://doi.org/10.1186/s12876-022-02159-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-022-02159-3